library(scran)
library(dplyr)
library(tidyr)
library(ggplot2)
library(scuttle)
library(scater)
library(DT)
library(patchwork)
library(SingleCellExperiment)
library(stringr)
library(gridExtra)data_viz
Data viz
Different ways to look at the preprocessed dataset
Preamble
Data
sce <- readRDS(file.path("..", "data", "sce_all_metadata_genes.rds"))
meta_dat <- read.csv(file.path("..", "data", "metadata.csv"),
sep = "\t", row.names = 1)
# major celltypes
sce$ct_broad <- sce$cell_type_name |> forcats::fct_collapse(
"capillary" = c("1 capillary1", "2 capillary2"),
"precollector" = c("3 precollector1", "4 precollector2"),
"collector" = c("5 collector"),
"valve" = c("6 valve"),
"prolieferative" = c("7 proliferative"))
#colours to correspond to shinycell
cList = list("cell_type_name" = c("#A6CEE3","#99CD91","#B89B74",
"#F06C45","#ED8F47","#825D99","#B15928"),
"tissue" = c("#A6CEE3","#F06C45","#B15928"),
"donor" = c("#A6CEE3","#99CD91","#B89B74",
"#F06C45","#ED8F47","#825D99","#B15928"))
names(cList[["cell_type_name"]]) <- c("1 capillary1","2 capillary2","3 precollector1","4 precollector2","5 collector","6 valve","7 proliferative")
names(cList[["tissue"]]) <- c("fat","mixed","skin")
names(cList[["donor"]]) <- c("1.0","2.0","3.0","4.0","5.0","6.0","7.0")cells per ct
table(sce$tissue)
fat mixed skin
7300 4705 9369 table(sce$donor)
1.0 2.0 3.0 4.0 5.0 6.0 7.0
3902 2773 1247 631 4705 3647 4469 table(sce$cell_type_name)
1 capillary1 2 capillary2 3 precollector1 4 precollector2 5 collector
6023 1156 5407 6041 1105
6 valve 7 proliferative
1555 87 table(sce$ct_broad)
capillary precollector collector valve prolieferative
7179 11448 1105 1555 87 table(sce$ct_broad, sce$tissue)
fat mixed skin
capillary 2206 1412 3561
precollector 3999 2663 4786
collector 613 249 243
valve 449 371 735
prolieferative 33 10 44table(sce$cell_type_name, sce$tissue)
fat mixed skin
1 capillary1 1748 1167 3108
2 capillary2 458 245 453
3 precollector1 1750 1163 2494
4 precollector2 2249 1500 2292
5 collector 613 249 243
6 valve 449 371 735
7 proliferative 33 10 44Data viz
Umap split by tissue
sce_fat <- sce[,sce$tissue %in% "fat"]
sce_skin <- sce[,sce$tissue %in% "skin"]
sce_mixed <- sce[,sce$tissue %in% "mixed"]
p1 <- plotReducedDim(sce_fat, dimred="umap",
colour_by="cell_type_name",
point_size = 0.8) +
ggtitle("fat") +
scale_color_manual(values = cList[["cell_type_name"]])Scale for colour is already present.
Adding another scale for colour, which will replace the existing scale.p2 <- plotReducedDim(sce_skin, dimred="umap",
colour_by="cell_type_name",
point_size = 0.8) +
ggtitle("skin") +
scale_color_manual(values = cList[["cell_type_name"]])Scale for colour is already present.
Adding another scale for colour, which will replace the existing scale.p3 <- plotReducedDim(sce_mixed, dimred="umap",
colour_by="cell_type_name",
point_size = 0.8) +
ggtitle("mixed") +
scale_color_manual(values = cList[["cell_type_name"]])Scale for colour is already present.
Adding another scale for colour, which will replace the existing scale.wrap_plots(list("skin" = p2,
"mixed" = p3,
"fat" = p1), nrow = 1) +
plot_layout(guides = "collect") &
theme(legend.position = "bottom")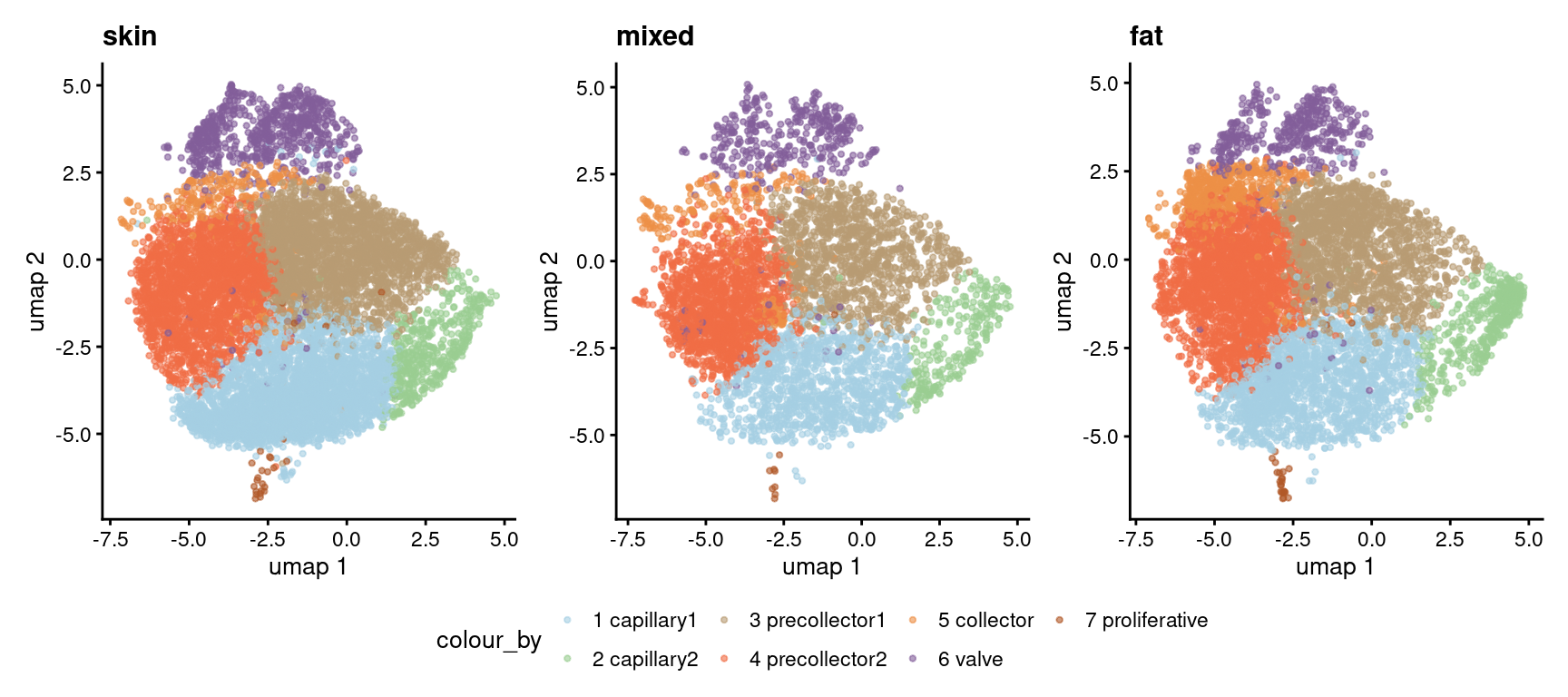
Umap before after integration
no_int <- calculateUMAP(reducedDims(sce)[["logcounts.scaled.pca"]],transposed = T)
reducedDims(sce)[["logcounts.scaled.umap"]] <- no_int
p1 <- plotReducedDim(sce, dimred="umap", colour_by="donor", point_size = 0.8) +
ggtitle("after") +
scale_color_manual(values = cList[["donor"]])Scale for colour is already present.
Adding another scale for colour, which will replace the existing scale.p2 <- plotReducedDim(sce, dimred="logcounts.scaled.umap", colour_by="donor", point_size = 0.8) +
ggtitle("before") +
scale_color_manual(values = cList[["donor"]])Scale for colour is already present.
Adding another scale for colour, which will replace the existing scale.wrap_plots(list("before" = p2,
"after" = p1), nrow = 1) +
plot_layout(guides = "collect") &
theme(legend.position = "bottom")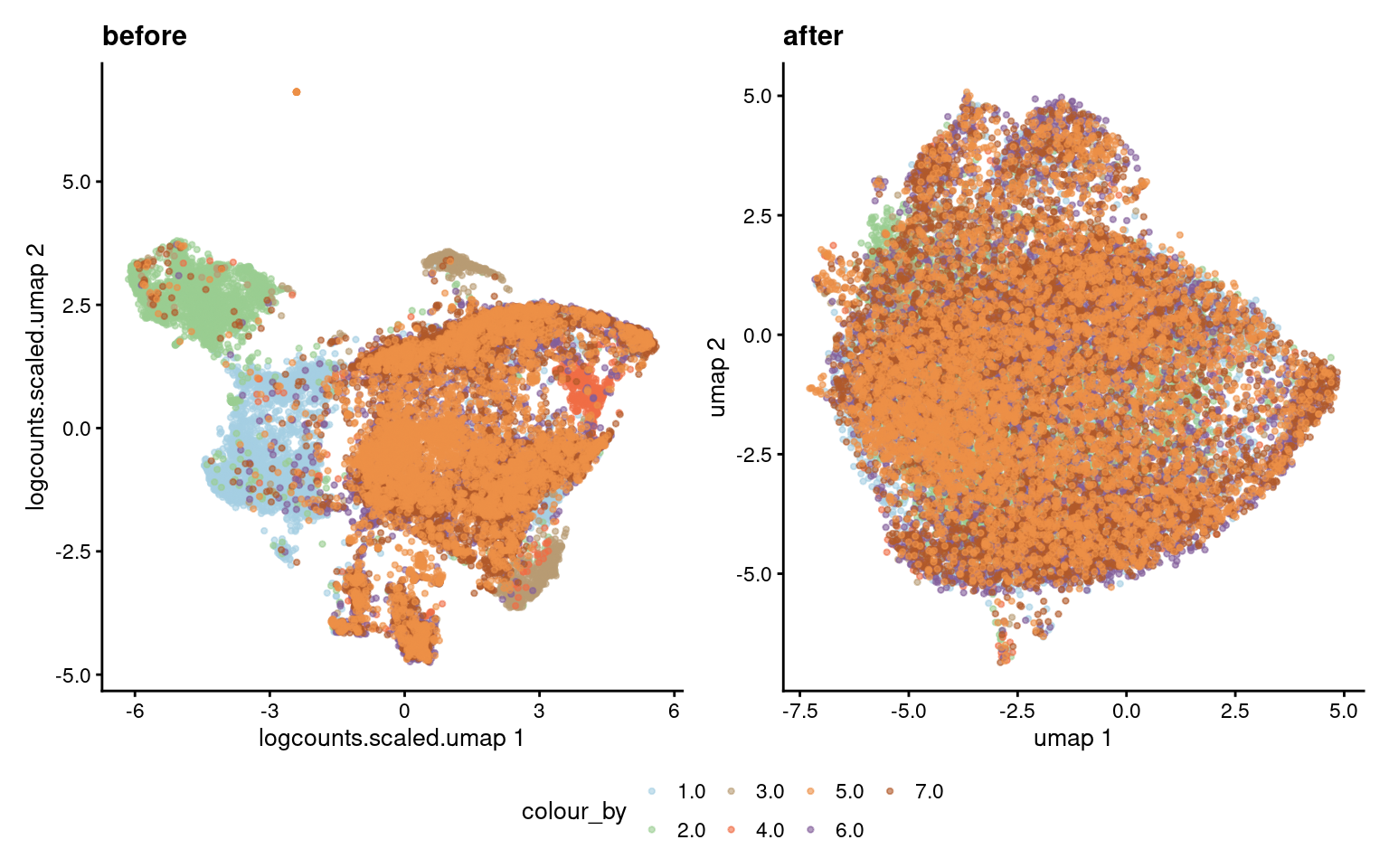
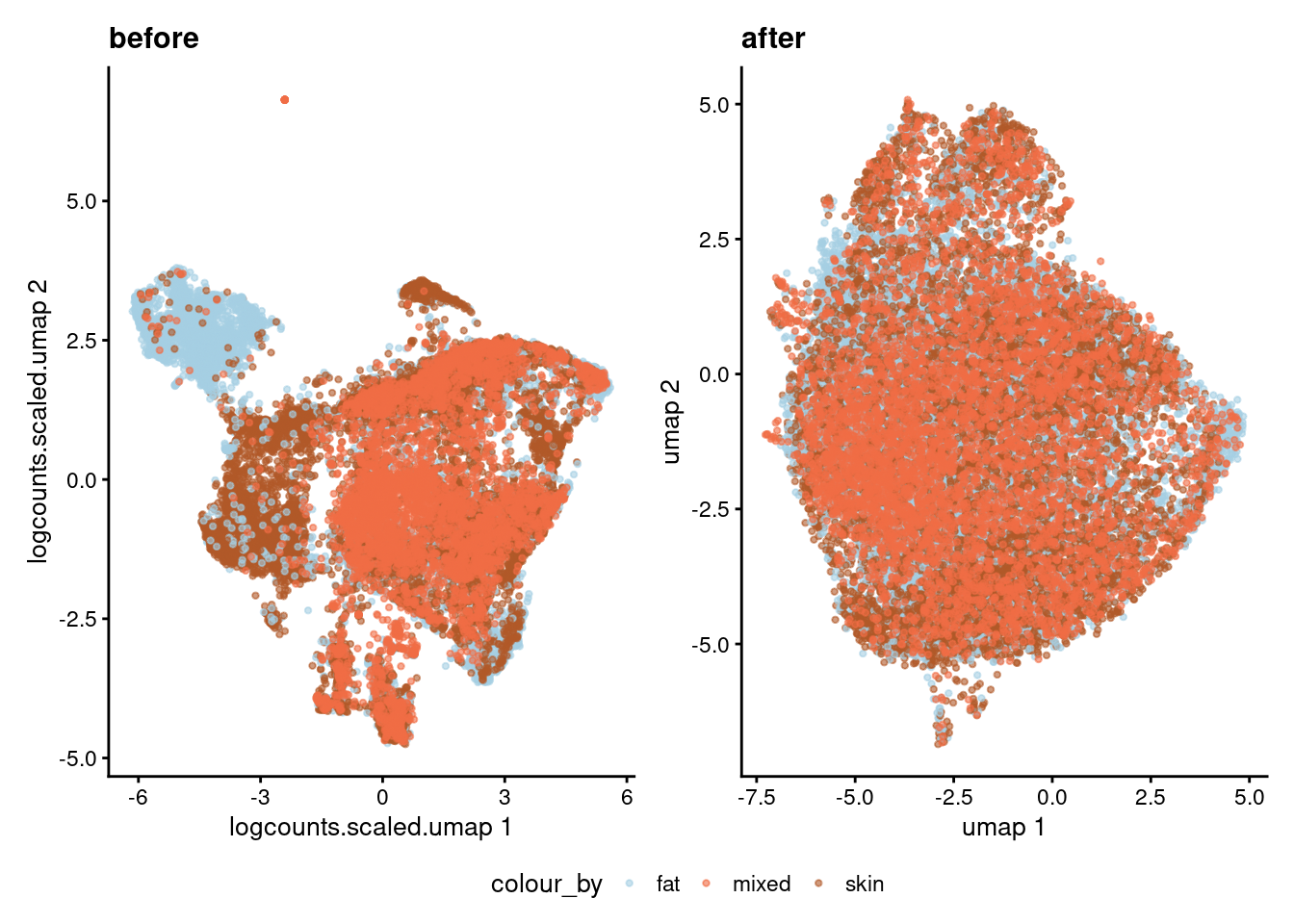
coloured by tissue
p1 <- plotReducedDim(sce, dimred="umap", colour_by="tissue", point_size = 0.8) +
ggtitle("after") +
scale_color_manual(values = cList[["tissue"]])Scale for colour is already present.
Adding another scale for colour, which will replace the existing scale.p2 <- plotReducedDim(sce, dimred="logcounts.scaled.umap", colour_by="tissue", point_size = 0.8) +
ggtitle("before") +
scale_color_manual(values = cList[["tissue"]])Scale for colour is already present.
Adding another scale for colour, which will replace the existing scale.wrap_plots(list("before" = p2,
"after" = p1), nrow = 1) +
plot_layout(guides = "collect") &
theme(legend.position = "bottom")