library(scran)
library(dplyr)
library(tidyr)
library(ggplot2)
library(edgeR)
library(stringr)
library(SingleCellExperiment)pb_de
Differential expression analysis
Pseudobulk DE analysis on major cell types using edgeR
Preamble
Data
sce <- readRDS(file.path("..", "data", "sce_all_metadata_genes.rds"))
meta_dat <- read.csv(file.path("..", "data", "metadata.csv"),
sep = "\t", row.names = 1)
# major celltypes
sce$ct_broad <- sce$cell_type_name |> forcats::fct_collapse(
"capillary" = c("1 capillary1", "2 capillary2"),
"precollector" = c("3 precollector1", "4 precollector2"),
"collector" = c("5 collector"),
"valve" = c("6 valve"),
"prolieferative" = c("7 proliferative"))
# major celltypes
sce$sub_cell_type <- sce$cell_type_name |> forcats::fct_collapse(
"capillary1" = "1 capillary1",
"capillary2" = "2 capillary2",
"precollector1" = "3 precollector1",
"precollector2" = "4 precollector2",
"collector" = "5 collector",
"valve" = "6 valve",
"prolieferative" = "7 proliferative")
# filter sample
sce <- sce[, !sce$donor %in% c("2.0", "5.0")]
sce$donor <- sce$donor |> droplevels()
sce$tissue <- sce$tissue |> droplevels()
# mean_expr
mean_expr_fat <- rowMeans(logcounts(sce[,sce$tissue %in% "fat"]))
mean_expr_skin <- rowMeans(logcounts(sce[,sce$tissue %in% "skin"]))Pseudobulk DE
summed <- aggregateAcrossCells(sce,
id=colData(sce)[,c("ct_broad", "donor", "tissue")])
# filter min cells
summed.filt <- summed[,summed$ncells >= 10]
print(table(summed.filt$tissue, summed.filt$ct_broad))
capillary precollector collector valve prolieferative
fat 5 5 3 4 0
skin 5 5 4 5 3# de
design <- model.matrix(~ donor + tissue, as.data.frame(colData(summed.filt)))
de.results <- pseudoBulkDGE(summed.filt,
label=summed.filt$ct_broad,
design=~donor + tissue,
coef="tissueskin",
condition=summed.filt$tissue
)
is.de <- decideTestsPerLabel(de.results, threshold=0.05)
summarizeTestsPerLabel(is.de) -1 0 1 NA
capillary 286 8886 127 9328
collector 5 3559 1 15062
precollector 23 10229 37 8338
valve 9 6550 10 12058Pseudobulk subcelltypes
summed <- aggregateAcrossCells(sce,
id=colData(sce)[,c("sub_cell_type", "donor", "tissue")])
# filter min cells
summed.filt <- summed[,summed$ncells >= 10]
print(table(summed.filt$tissue, summed.filt$sub_cell_type))
capillary1 capillary2 precollector1 precollector2 collector valve
fat 5 4 5 5 3 4
skin 5 5 5 5 4 5
prolieferative
fat 0
skin 3# de
design <- model.matrix(~ donor + tissue, as.data.frame(colData(summed.filt)))
de_sub.results <- pseudoBulkDGE(summed.filt,
label=summed.filt$sub_cell_type,
design=~donor + tissue,
coef="tissueskin",
condition=summed.filt$tissue
)
is.de <- decideTestsPerLabel(de_sub.results, threshold=0.05)
summarizeTestsPerLabel(is.de) -1 0 1 NA
capillary1 13 8529 23 10062
capillary2 41 4795 40 13751
collector 5 3559 1 15062
precollector1 21 9317 23 9266
precollector2 8 7692 8 10919
valve 9 6550 10 12058#save results
lapply(names(de_sub.results), function(nam){
saveRDS(de_sub.results[[nam]], file.path("..", "out", "de", "subcelltypes", paste0("pb_", nam, ".rds")))
write.csv(de_sub.results[[nam]], file.path("..", "out", "de", "subcelltypes", paste0("pb_", nam, ".csv")))
})[[1]]
NULL
[[2]]
NULL
[[3]]
NULL
[[4]]
NULL
[[5]]
NULL
[[6]]
NULLPseudobulk DE all
summed_all <- aggregateAcrossCells(sce,
id=colData(sce)[,c("donor", "tissue")])
# filter min cells
summed_all.filt <- summed_all[,summed_all$ncells >= 10]
table(summed_all.filt$tissue)
fat skin
5 5 # de
design <- model.matrix(~ donor + tissue, as.data.frame(colData(summed_all.filt)))
dgl <- DGEList(counts(summed_all.filt))
keep <- filterByExpr(dgl, design)
dgl <- dgl[keep, , keep.lib.sizes=FALSE]
dgl <- calcNormFactors(dgl)
dgl <- estimateDisp(dgl, design)
fit <- glmQLFit(dgl, design)
de <- glmQLFTest(fit, coef="tissueskin")
tt <- topTags(de, n = Inf)$table
tt$full_gene_name <- rownames(tt)
tt$gene_symbol <- gsub("^.*\\.","",rownames(tt))
tt$meanExpr_fat <- mean_expr_fat[rownames(tt)]
tt$meanExpr_skin <- mean_expr_skin[rownames(tt)]
saveRDS(tt, file.path("..", "out", "de", "pb_all.rds"))
write.csv(tt, file.path("..", "out", "de", "pb_all.csv"))Differential abundance
sce_sub <- sce[,!sce$Sample %in% c("donor5_recovery", "donor2_fat")]
sce_sub$Sample <- sce_sub$Sample |> droplevels()
abundances <- table(sce_sub$cell_type_name, sce_sub$Sample)
abundances <- unclass(abundances)
extra.info <- colData(sce_sub)[match(colnames(abundances), sce_sub$Sample),]
y.ab <- DGEList(abundances, samples=extra.info)
design <- model.matrix(~factor(donor) + factor(tissue), y.ab$samples)
y.ab <- estimateDisp(y.ab, design, trend="none")
summary(y.ab$common.dispersion) Min. 1st Qu. Median Mean 3rd Qu. Max.
0.2145 0.2145 0.2145 0.2145 0.2145 0.2145 plotBCV(y.ab, cex=1)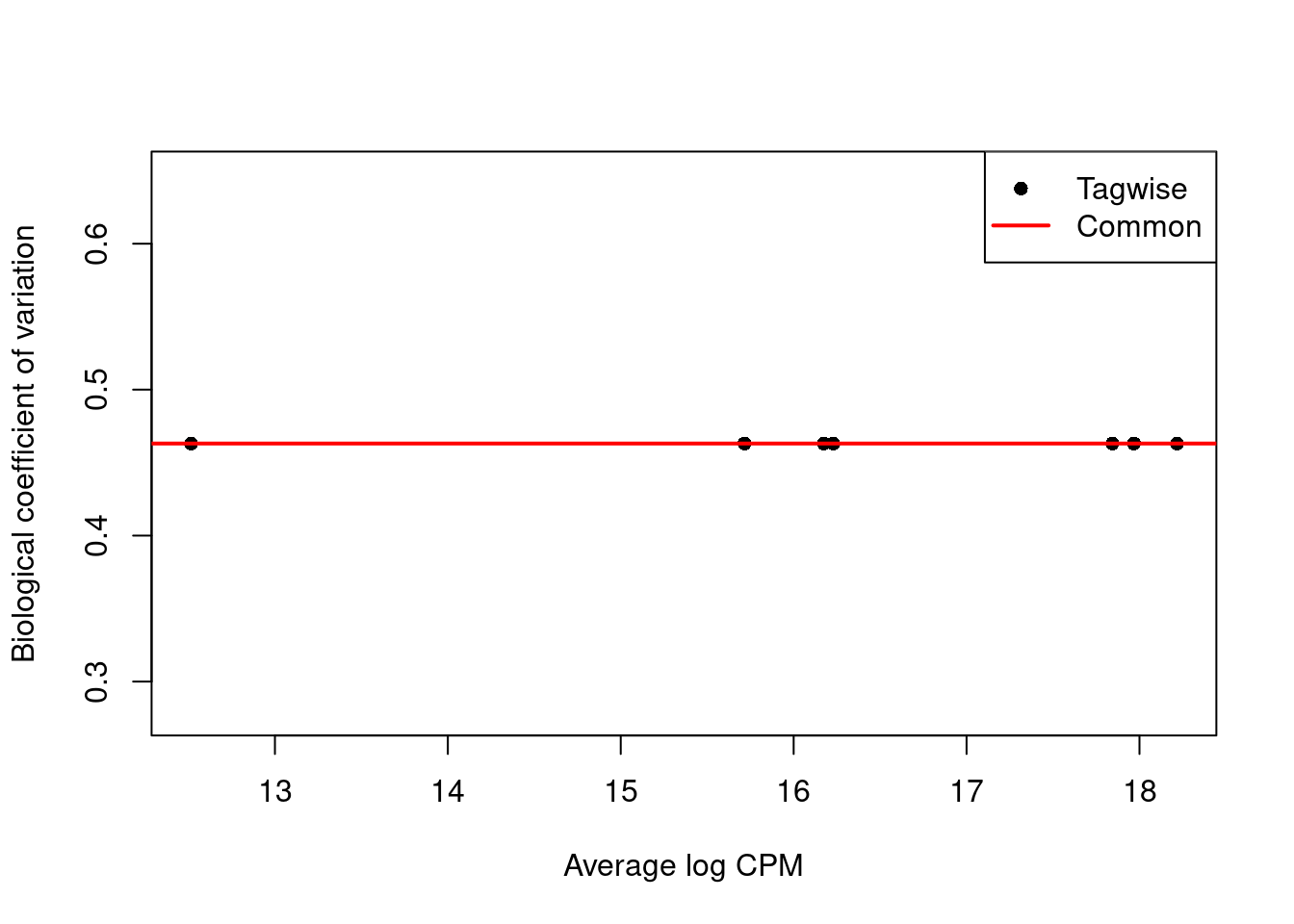
fit.ab <- glmQLFit(y.ab, design, robust=TRUE, abundance.trend=FALSE)
summary(fit.ab$var.prior)Length Class Mode
0 NULL NULL summary(fit.ab$df.prior) Min. 1st Qu. Median Mean 3rd Qu. Max.
116.4 116.4 116.4 116.4 116.4 116.4 plotQLDisp(fit.ab, cex=1)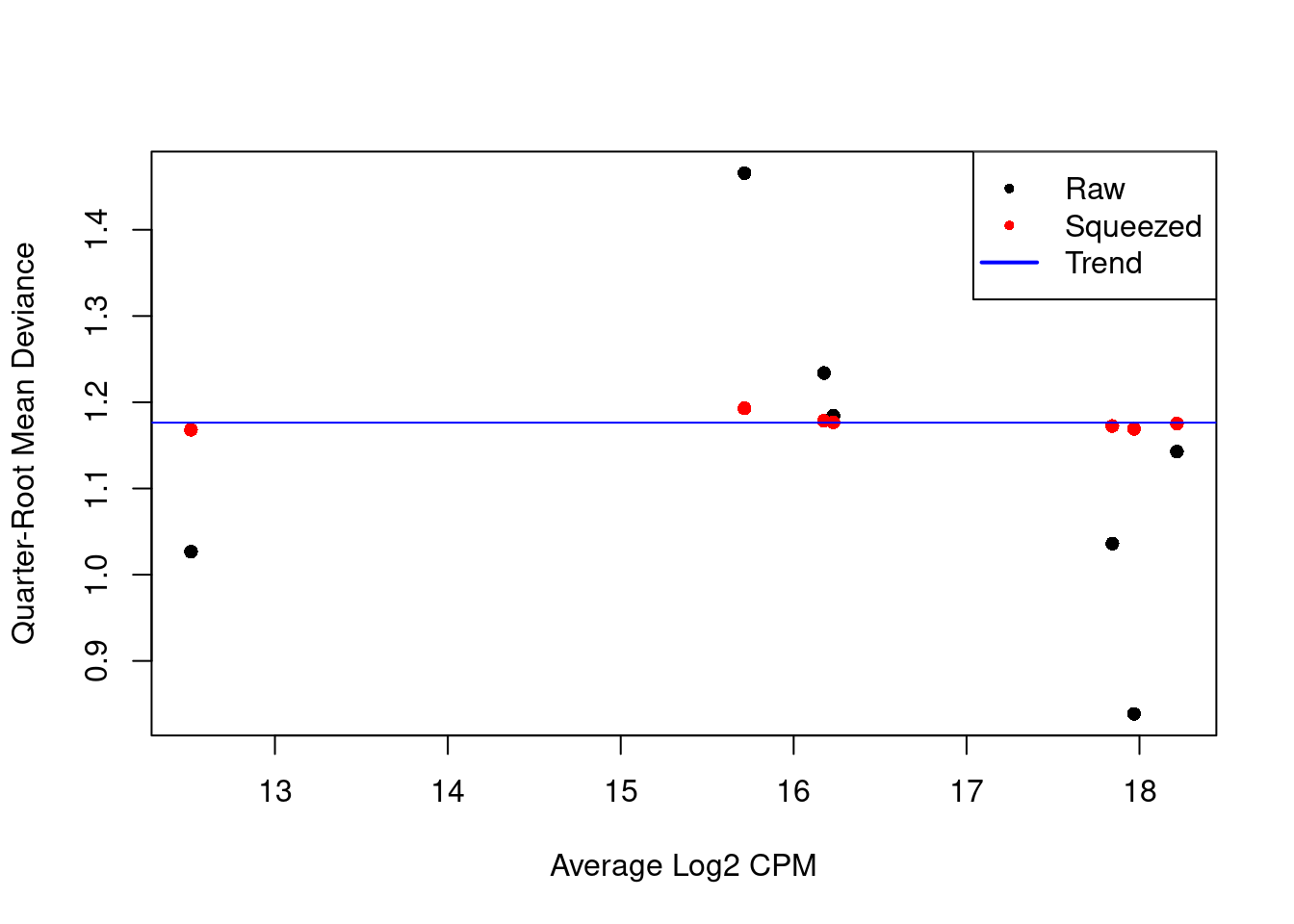
res <- glmQLFTest(fit.ab, coef=ncol(design))
summary(decideTests(res)) factor(tissue)skin
Down 0
NotSig 7
Up 0topTags(res)Coefficient: factor(tissue)skin
logFC logCPM F PValue FDR
5 collector -1.54832644 15.71539 8.369045854 0.007308034 0.05115624
1 capillary1 0.83551888 18.21660 4.076859927 0.053141232 0.18599431
4 precollector2 -0.71376716 17.84314 3.005225055 0.093996945 0.21932621
2 capillary2 -0.59024211 16.17506 1.681977784 0.205247728 0.35918352
6 valve 0.26641377 16.22953 0.343084253 0.562746204 0.78784469
7 proliferative 0.16178462 12.51469 0.036642640 0.849575826 0.92980460
3 precollector1 -0.03573641 17.96845 0.007900865 0.929804601 0.92980460