library(scran)
library(dplyr)
library(tidyr)
library(ggplot2)
library(scuttle)
library(scater)
library(pheatmap)
library(ComplexHeatmap)
library(UpSetR)
library(patchwork)
library(EnhancedVolcano)
library(SingleCellExperiment)
library(stringr)
library(gridExtra)vis_de
Visualize DE results
Preambel
Libraries
Helper functions
Code
# volcano plot function as defined in https://github.com/HelenaLC/TLS-Silina/blob/main/code/geo-02-differential.qmd
.volcano <- \(df, title, fdr = 0.05, lfc = 1, select_lab = NULL) {
EnhancedVolcano(df,
x = "logFC", y = "FDR",
FCcutoff = lfc, pCutoff = fdr,
selectLab = select_lab,
pointSize = 1.7, raster = TRUE,
title = title, subtitle = NULL,
lab = rownames(df), labSize = 4,
drawConnectors = TRUE, widthConnectors = 0.5) +
guides(col = guide_legend(override.aes = list(alpha = 1, size = 5))) +
theme_bw(9) + theme(
aspect.ratio = 1,
legend.title = element_blank(),
panel.grid.minor = element_blank())
}Data objects
pb_de_files <- list.files("../out/de/20231024", pattern = ".csv", full.names = T)
pb_de_names <- list.files("../out/de/20231024", pattern = ".csv", full.names = F)
pb_de <- lapply(pb_de_files, read.csv, row.names=1)
names(pb_de) <- gsub("\\.csv","",pb_de_names)
names(pb_de)[1] "cap_pseudo_bulk" "coll_pseudo_bulk" "DEA"
[4] "precoll_pseudo_bulk" "valves_pseudo_bulk" pb_all <- readRDS("../out/de/pb_all.rds")
pb_all$gene_symbol[duplicated(pb_all$gene_symbol)] <- pb_all$full_gene_name[duplicated(pb_all$gene_symbol)]
rownames(pb_all) <- pb_all$gene_symbol
pb_de[["all"]] <- pb_all
pb_de <- pb_de[-which(names(pb_de) %in% "DEA")]
pb_valve <- readRDS("../out/de/valve2_vs_1.rds")
pb_cap1 <- readRDS("../out/de/subcelltypes/pb_capillary1.rds")
pb_cap2 <- readRDS("../out/de/subcelltypes/pb_capillary1.rds")
pb_precol1 <- readRDS("../out/de/subcelltypes/pb_precollector1.rds")
pb_precol2 <- readRDS("../out/de/subcelltypes/pb_precollector2.rds")
pb_sub <- list("capillary1" = pb_cap1,
"capillary2" = pb_cap2,
"precollector1" = pb_precol1,
"precollector2" = pb_precol2)
pb_sub <- lapply(pb_sub, function(pb_res){
pb_res$full_gene_name <- rownames(pb_res)
pb_res$gene_symbol <- gsub("^.*\\.","",rownames(pb_res))
pb_res$gene_symbol[duplicated(pb_res$gene_symbol)] <-
pb_res$full_gene_name[duplicated(pb_res$gene_symbol)]
rownames(pb_res) <- pb_res$gene_symbol
pb_res
})
sce <- readRDS(file.path("..", "data", "sce_all_metadata_genes.rds"))
sce <- sce[, !sce$donor %in% c("2.0", "5.0")]
sce$donor <- sce$donor |> droplevels()
sce$tissue <- sce$tissue |> droplevels()
#list to map de names to cell type names
ct_nam <- list("cap_pseudo_bulk" = c("capillary"),
"precoll_pseudo_bulk" = c("precollector"),
"valves_pseudo_bulk" = c("valve"))
ct_sub_nam <- list("capillary1" = "1 capillary1",
"capillary2" = "2 capillary2",
"precollector1" = "3 precollector1",
"precollector2" = "4 precollector2")
sce$ct_broad <- sce$cell_type_name |> forcats::fct_collapse(
"capillary" = c("1 capillary1", "2 capillary2"),
"precollector" = c("3 precollector1", "4 precollector2"),
"collector" = c("5 collector"),
"valve" = c("6 valve"),
"prolieferative" = c("7 proliferative"))
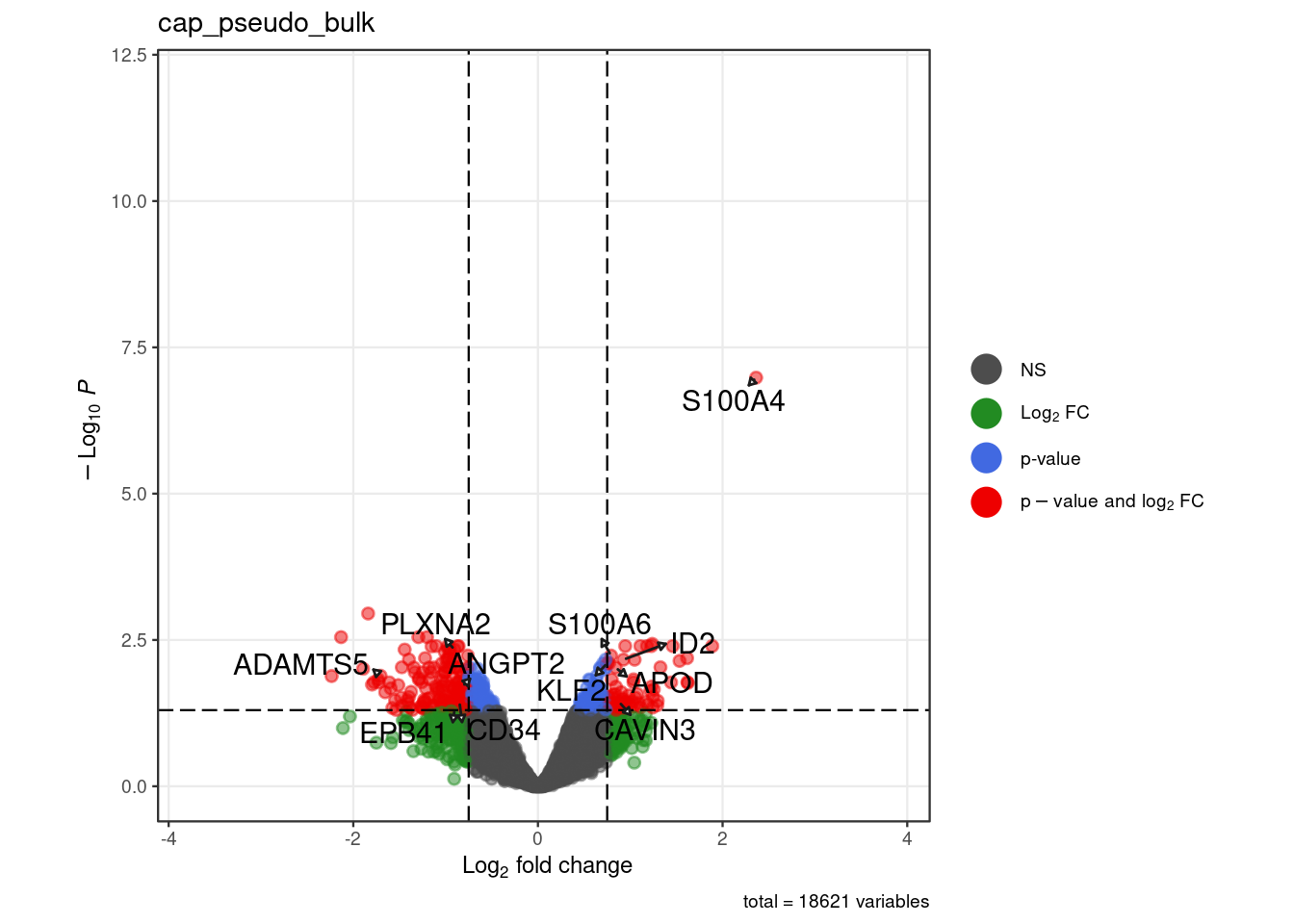
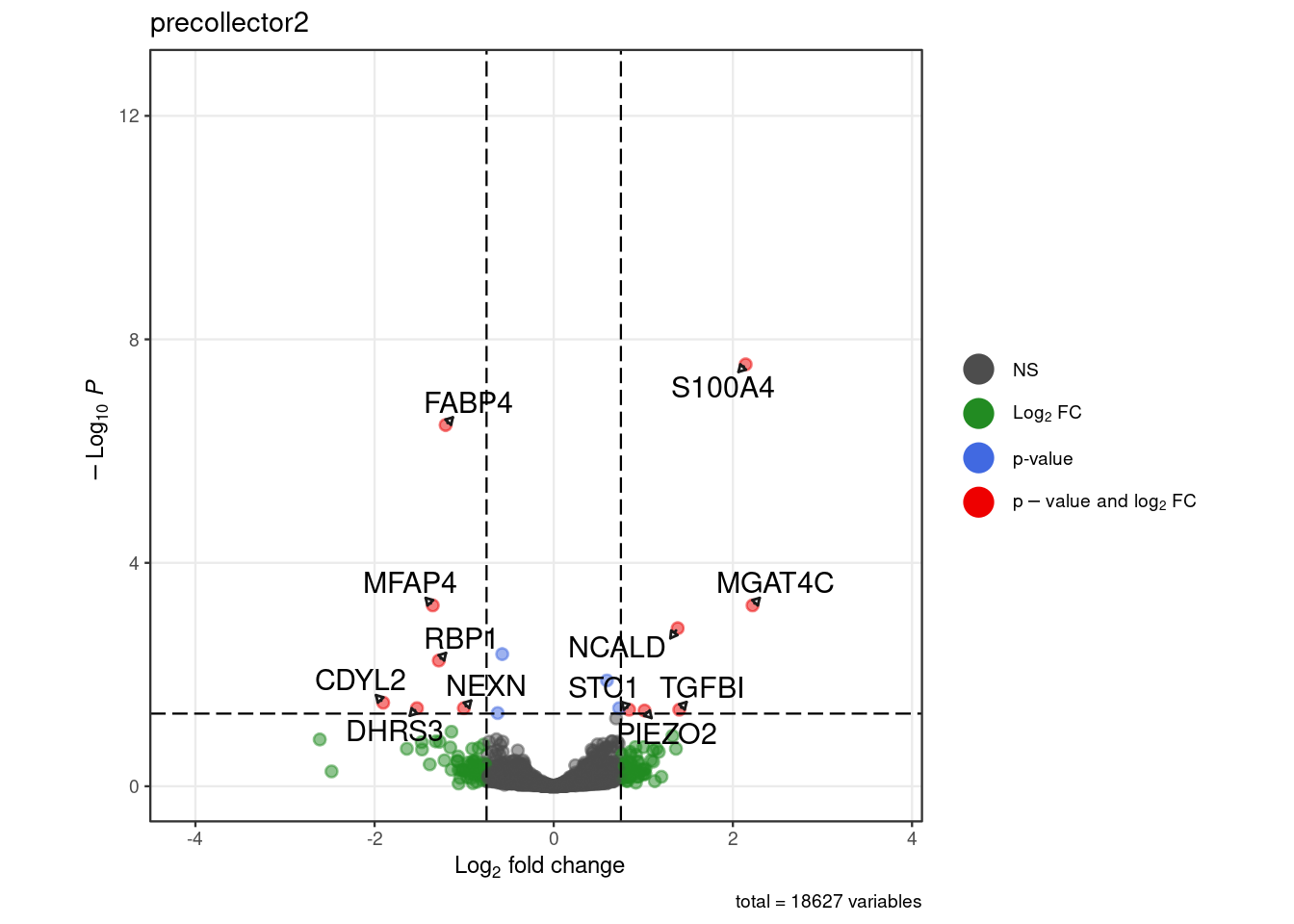
label_list <- list("cap_pseudo_bulk" = c("APOD","S100A4", "S100A6", "KLF2", "CAVIN3", "ID2",
"ADAMTS5", "PLXNA2", "EPB41", "CD34",
"ANGPT2"),
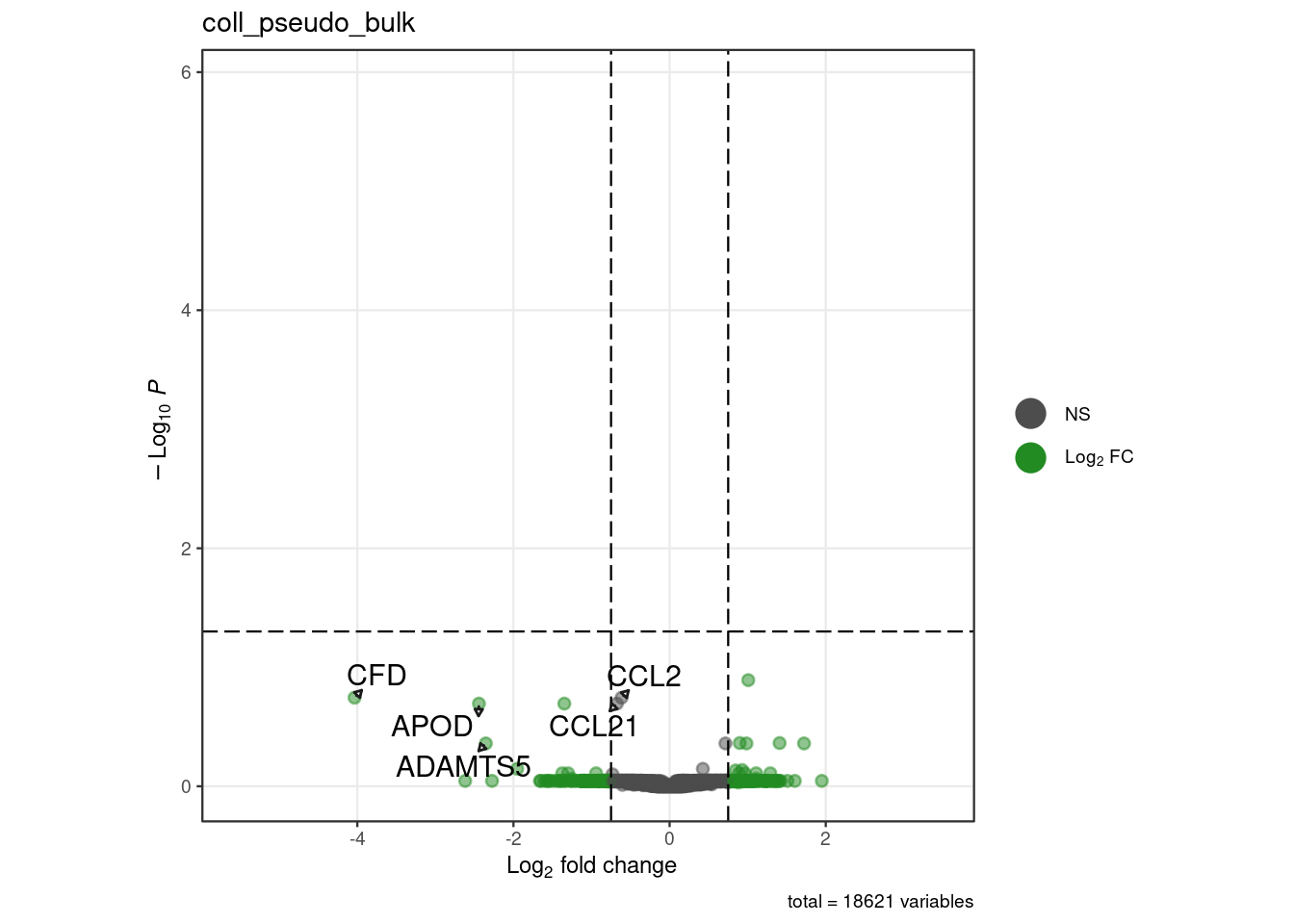
"coll_pseudo_bulk" = c("CFD","APOD", "ADAMTS5", "CCL2", "CCL21"),
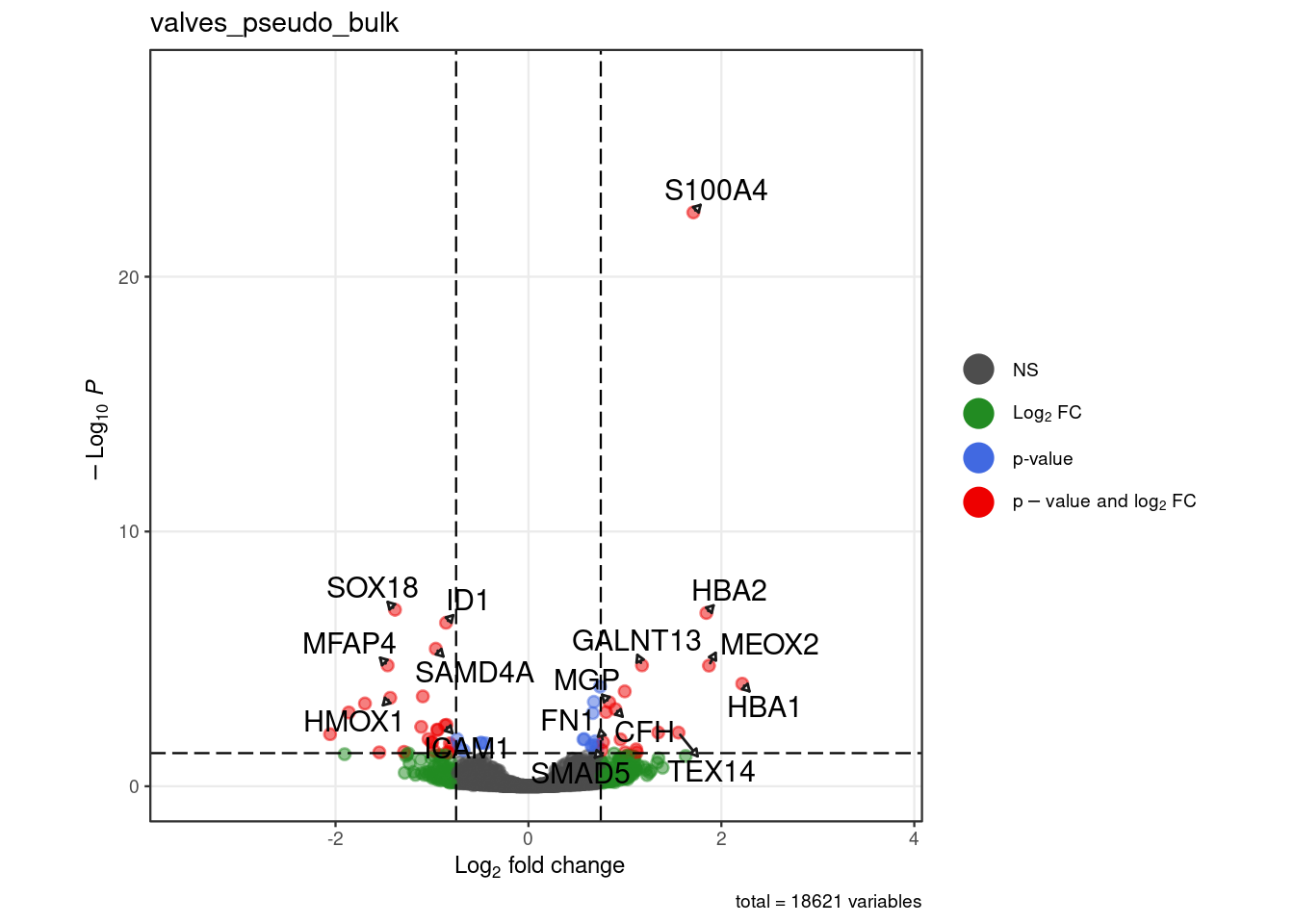
"valves_pseudo_bulk" = c("ICAM1", "S100A4","SOX18","ID1","SAMD4A",
"MFAP4","HMOX1","HBA2","MEOX2","HBA1","TEX14","SMAD5","FN1",
"GALNT13","CFH","MGP"),
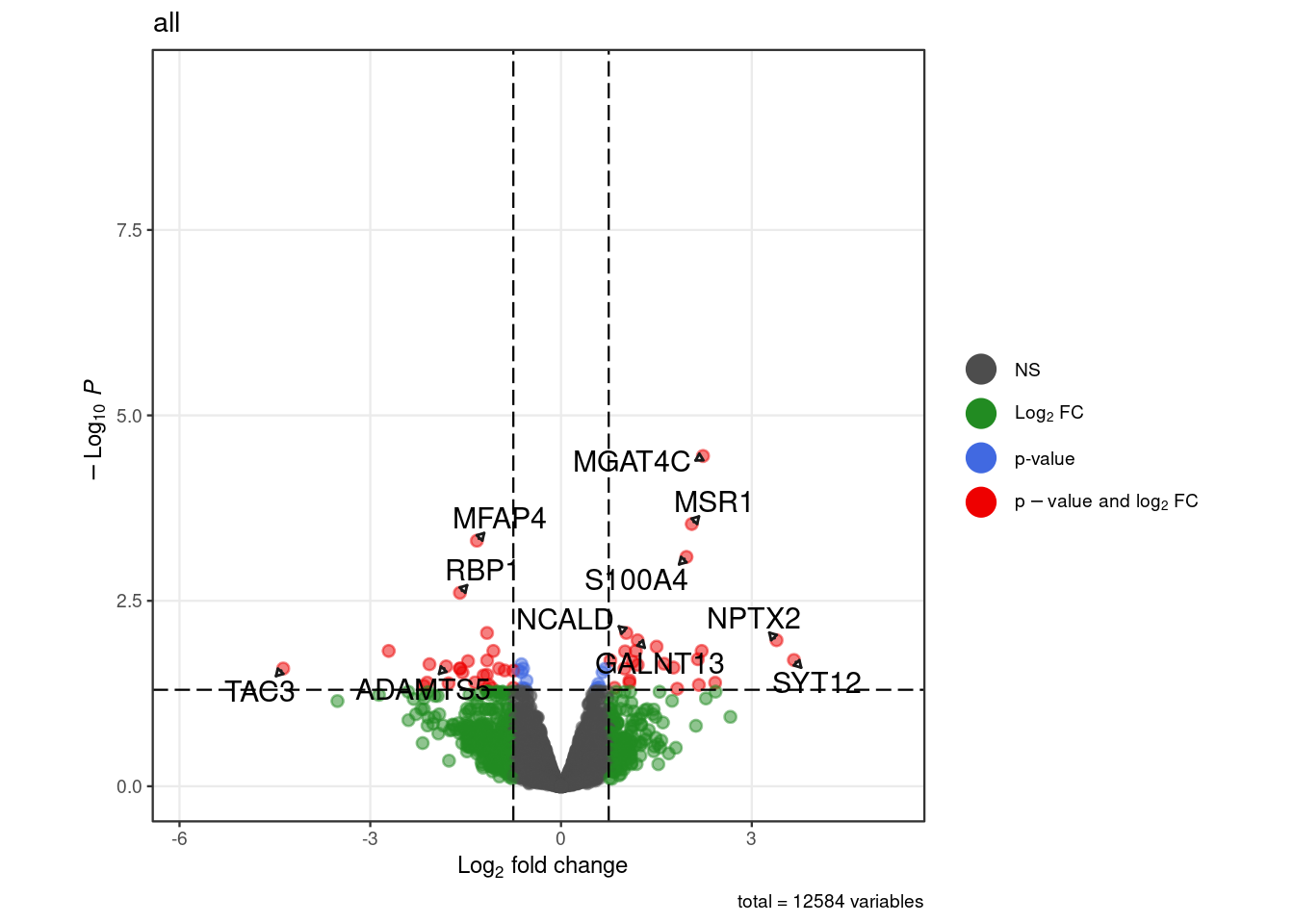
"all" = c("S100A4", "ADAMTS5", "COL5a2", "TAC3","RBP1","MFAP4","MGAT4C", "MSR1",
"NCALD","GALNT13","NPTX2","SYT12"))
valve_lab = c("NEO1","ADAMTS1","ADAMTS6","CD24","STC1","GRP","CCDC3","ACKR3",
"RCAN1","SEMA3D","FGL2","SLC12A2","ADAMTS9","ENOX1","IGFBP2",
"SERPINB1","CLDN11","ANKRD55","SCG3","HGF","HBA2","ADM","ANGPT2",
"CLU","APOD","GJA4","FST")Visualizations
Volcano(s)
for (. in names(pb_de)) {
p <- .volcano(df = pb_de[[.]], title = ., fdr = 0.05, lfc = 0.75,
select_lab =label_list[[.]])
cat("####", ., "\n"); print(p); cat("\n\n")
}
dup_ind <-which(duplicated(pb_valve$gene_symbol))
pb_valve$gene_symbol[dup_ind] <- paste0(pb_valve$full_gene_name[dup_ind],
".", pb_valve$gene_symbol[dup_ind])
rownames(pb_valve) <- pb_valve$gene_symbol
p_val <- .volcano(df = pb_valve, title = "valve2 vs valve1", fdr = 0.05,
lfc = 0.75, select_lab = valve_lab)
cat("####", "valve subgroups", "\n"); print(p_val); cat("\n\n")
# subcelltypes
for (. in names(pb_sub)) {
p <- .volcano(df = pb_sub[[.]], title = ., fdr = 0.05, lfc = 0.75)
cat("####", ., "\n"); print(p); cat("\n\n")
}
Warning: ggrepel: 14 unlabeled data points (too many overlaps). Consider
increasing max.overlaps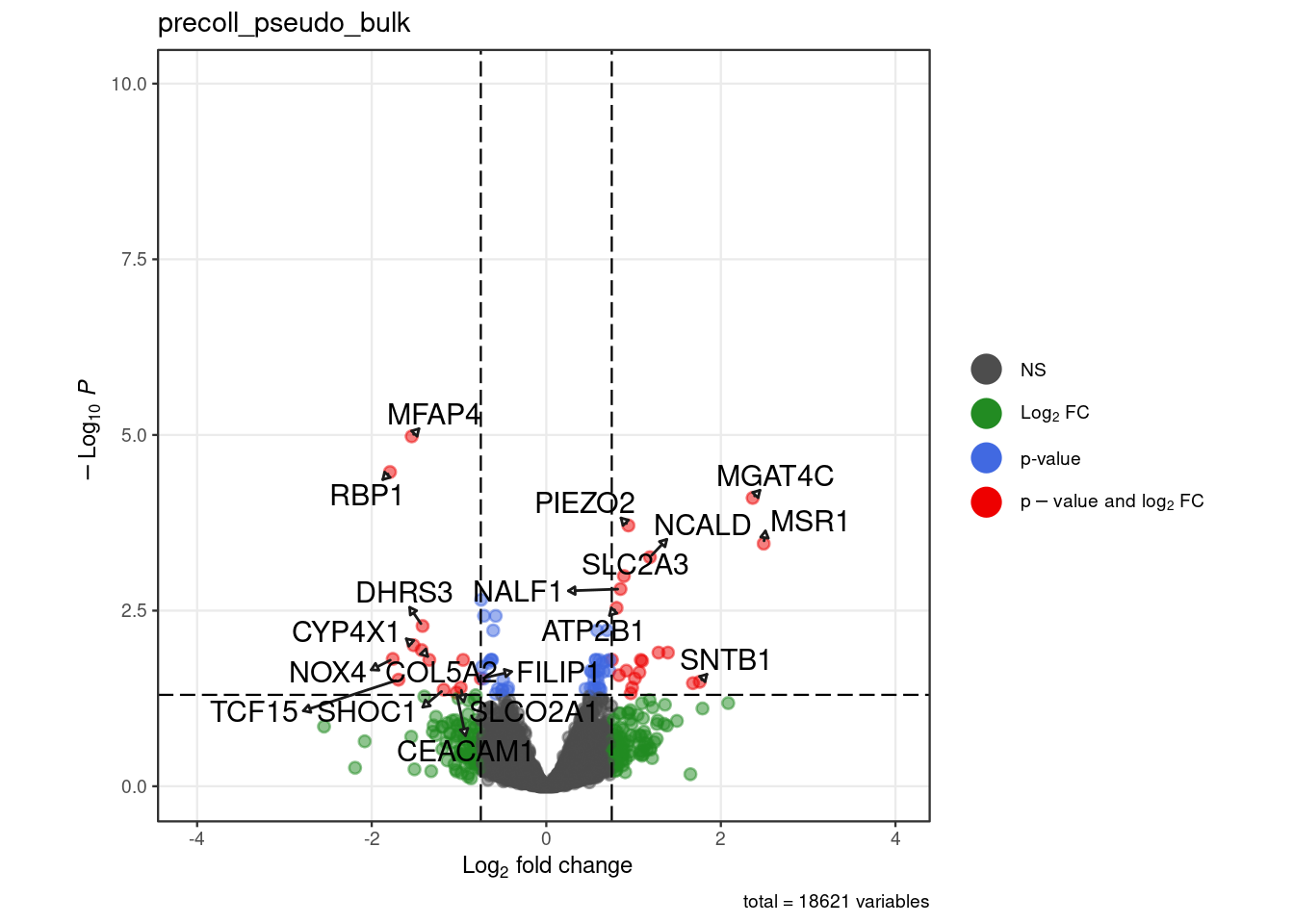
pb_valve$gene_symbol[duplicated(pb_valve$gene_symbol)] <- paste0(
pb_valve$full_gene_name[duplicated(pb_valve$gene_symbol)], ".",
pb_valve$gene_symbol[duplicated(pb_valve$gene_symbol)]
)
rownames(pb_valve) <- pb_valve$gene_symbol
p_val <- .volcano(df = pb_valve, title = "valve2 vs valve1", fdr = 0.05,
lfc = 0.75, select_lab = valve_lab)
cat("####", "valve subgroups", "\n"); print(p_val); cat("\n\n")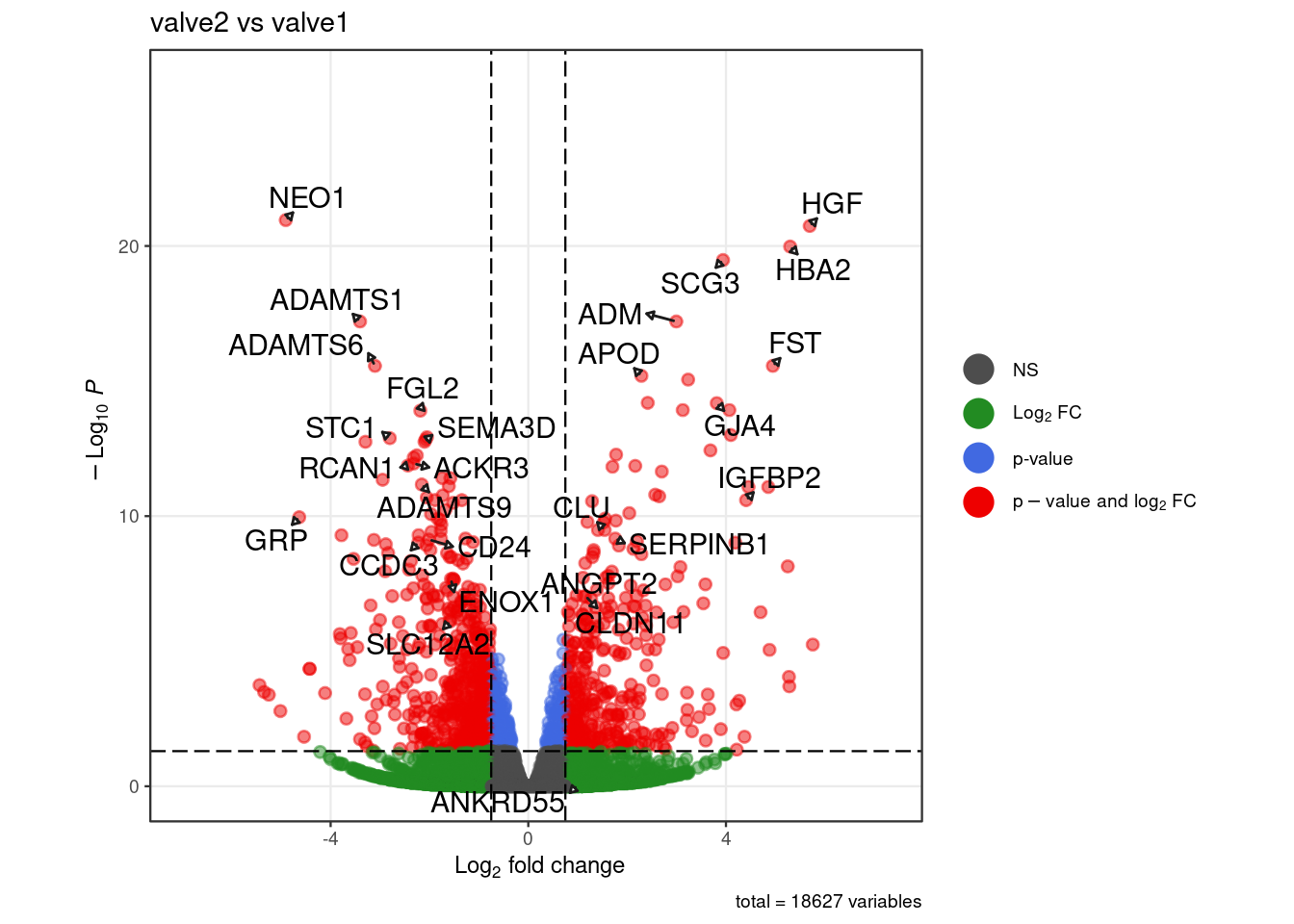
# subcelltypes
for (. in names(pb_sub)) {
p <- .volcano(df = pb_sub[[.]], title = ., fdr = 0.05, lfc = 0.75)
cat("####", ., "\n"); print(p); cat("\n\n")
}Warning: ggrepel: 9 unlabeled data points (too many overlaps). Consider
increasing max.overlaps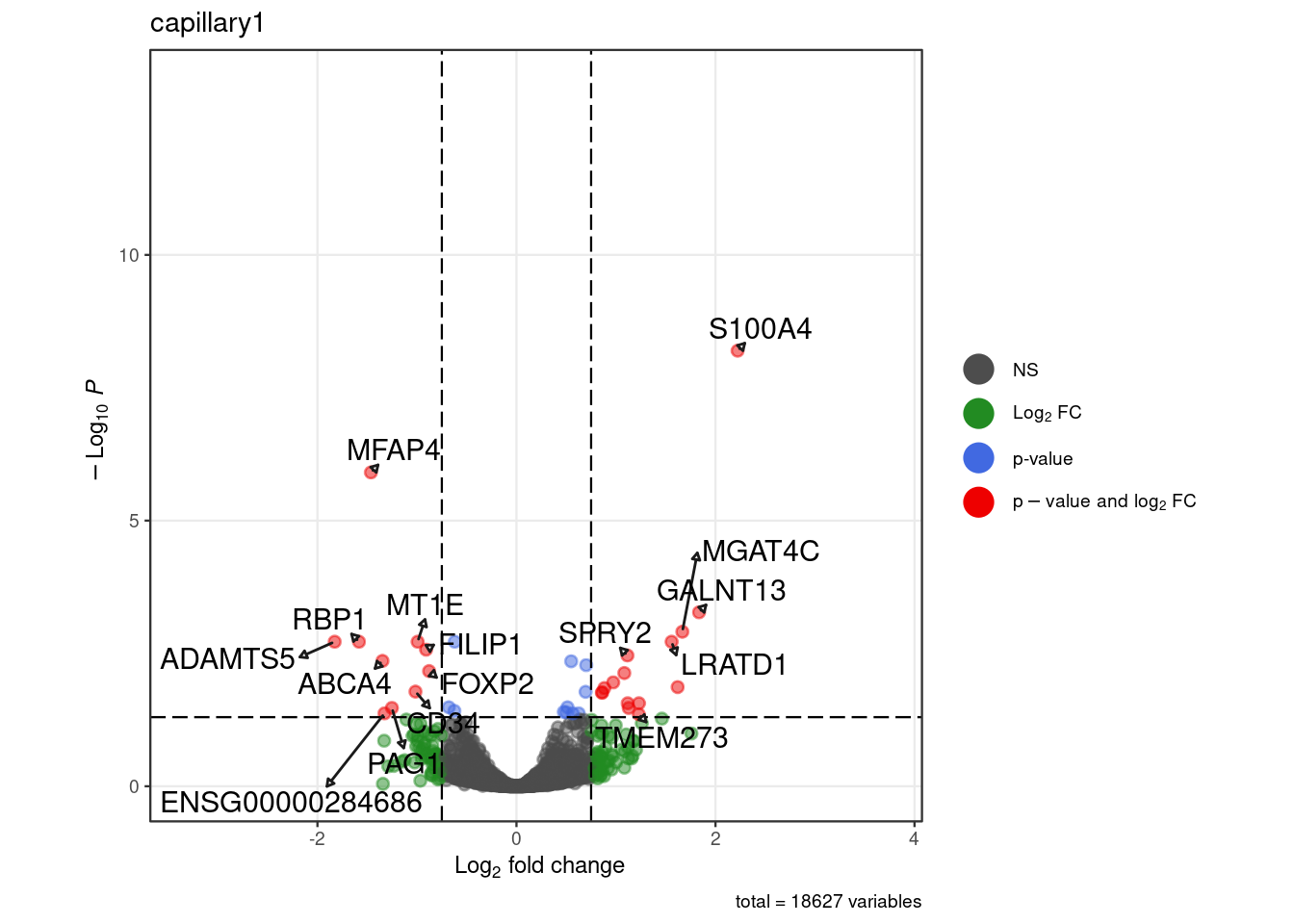
Warning: ggrepel: 9 unlabeled data points (too many overlaps). Consider
increasing max.overlaps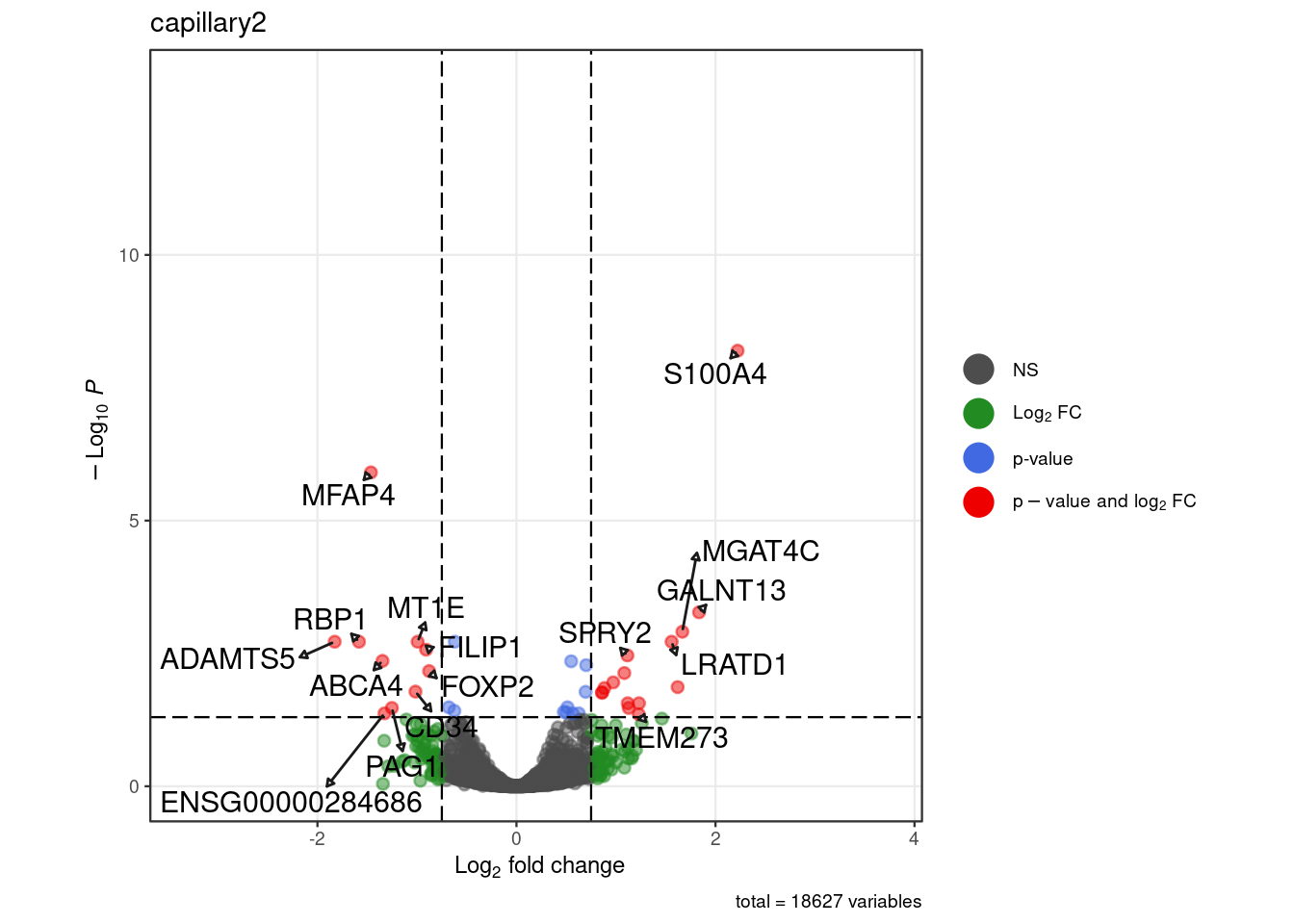
Warning: ggrepel: 12 unlabeled data points (too many overlaps). Consider
increasing max.overlaps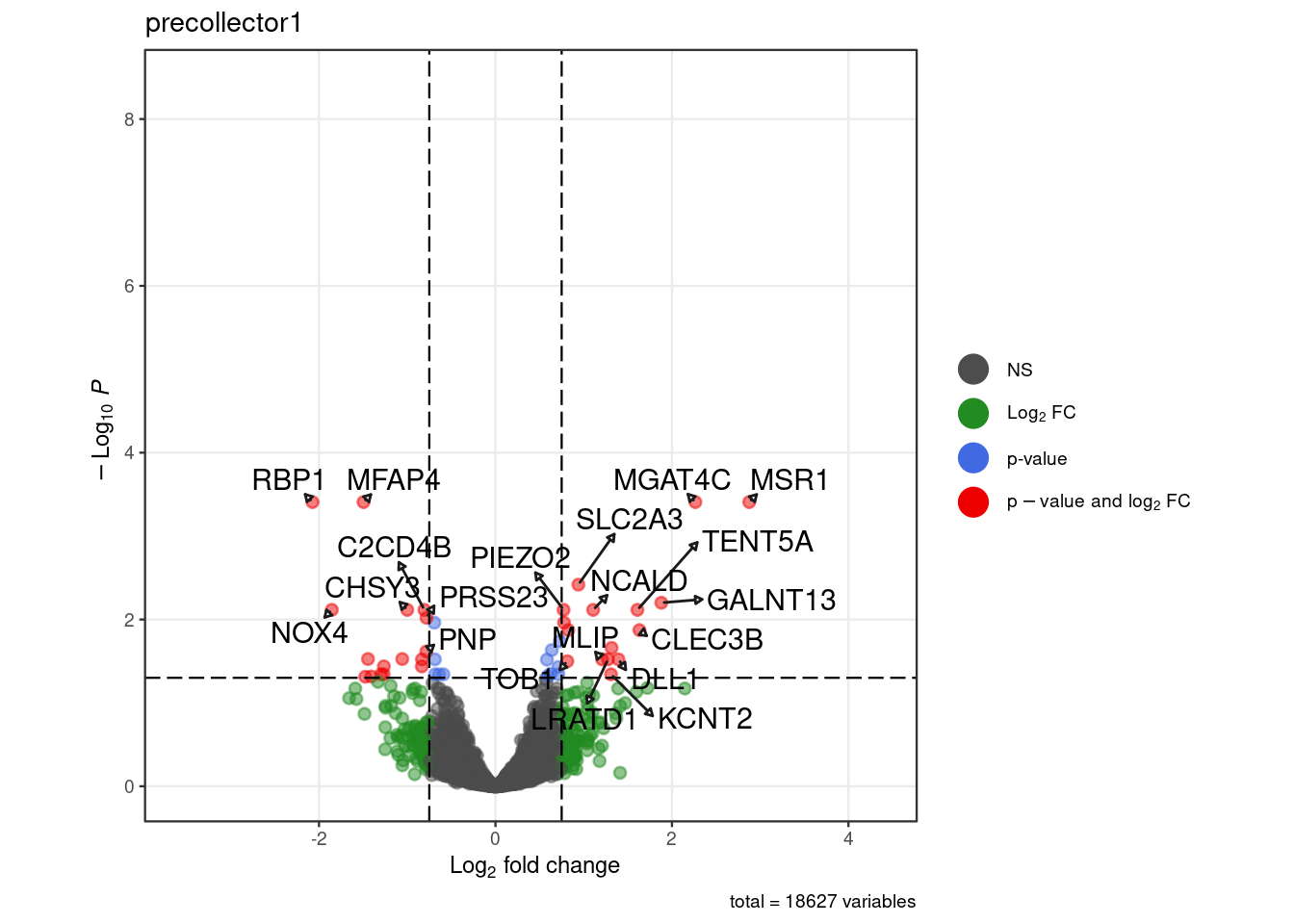
Pseudobulk Heatmap
Code
summed <- aggregateAcrossCells(sce,
id=colData(sce)[,c("tissue", "ct_broad", "donor")],
use.assay.type = "counts")
summed <- summed[,summed$ncells >= 10]
rownames(summed) <- gsub("^.*\\.","",rownames(summed))
# subset res to de-genes
de_list <- lapply(pb_de, function(.){
top <- as.data.frame(.) |>
filter(FDR < 0.01) |>
slice_max(abs(logFC), n = 20)
})
drop_ind <- which(sapply(de_list, nrow) < 1)
drop_ind2 <- which(names(de_list) %in% c("DEA", "all"))
de_gen_list <- de_list[-c(drop_ind, drop_ind2)]
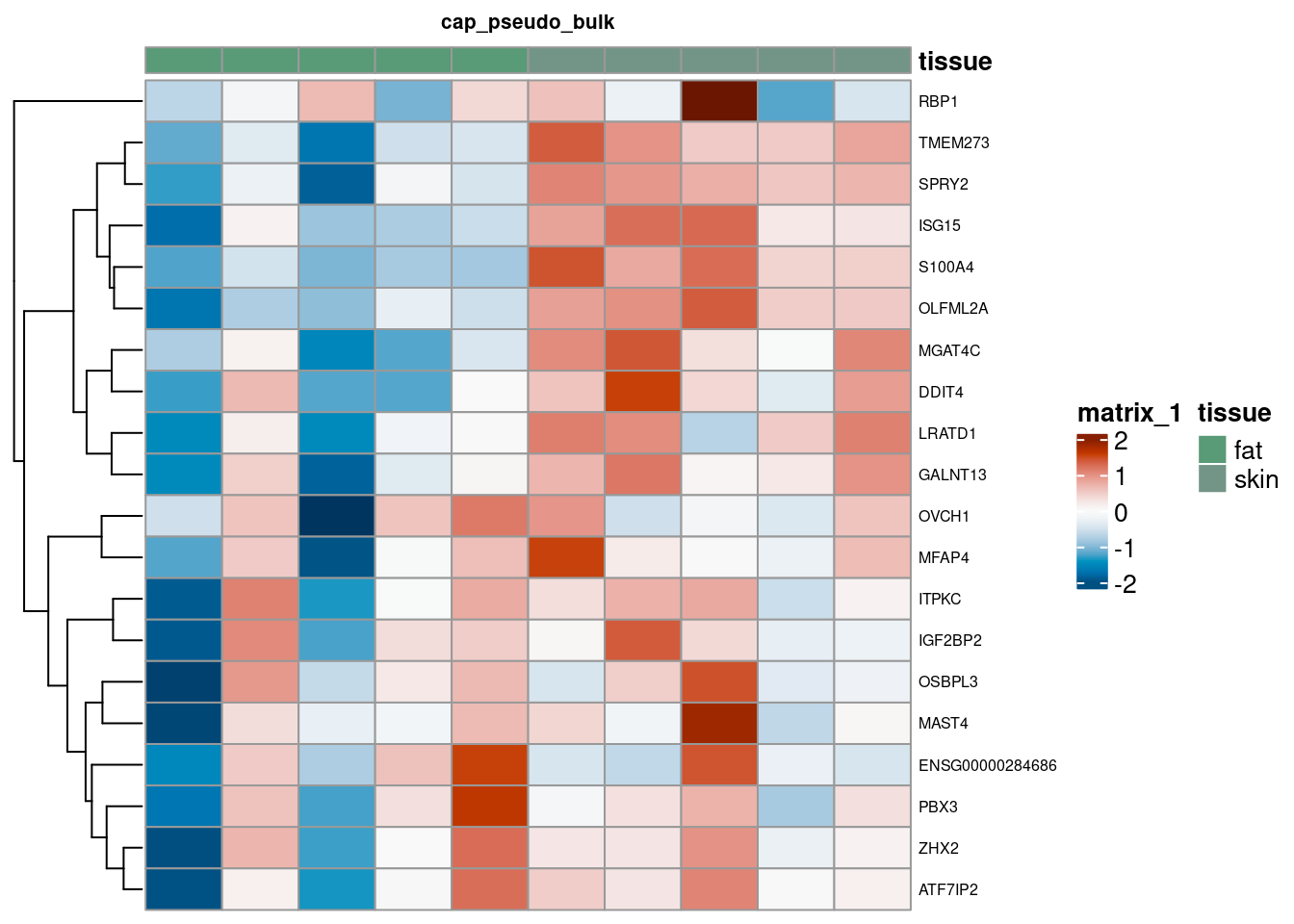
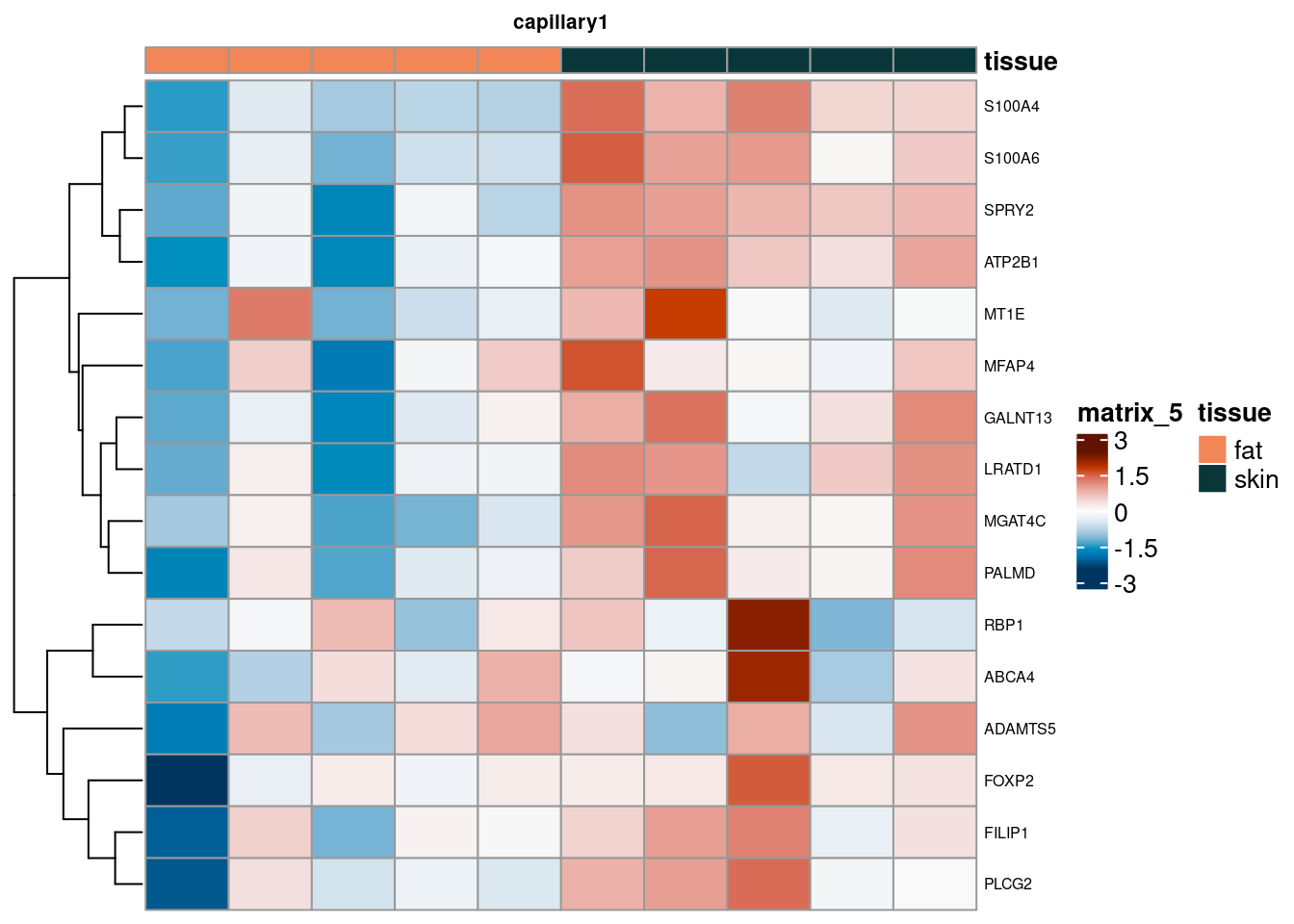
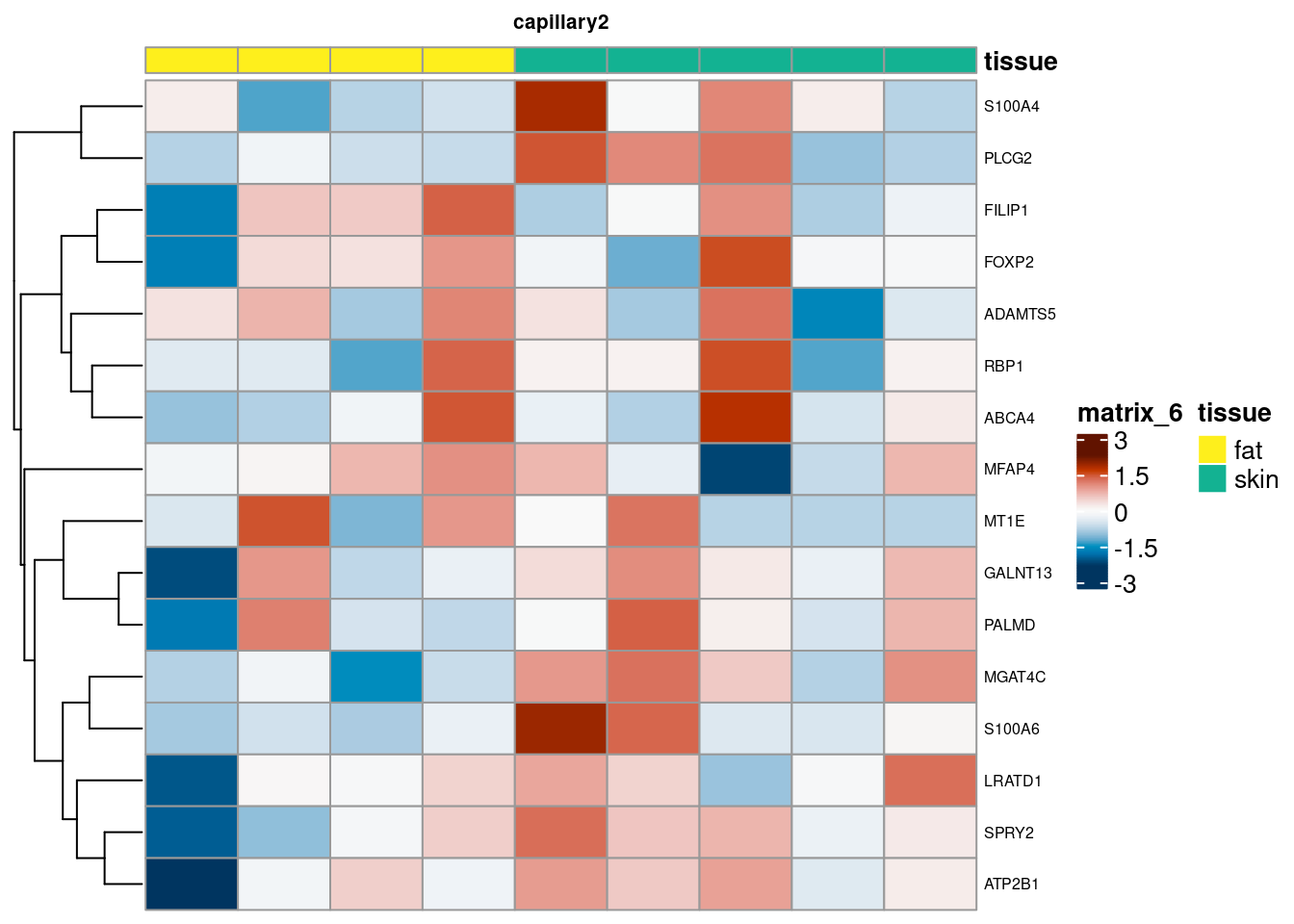
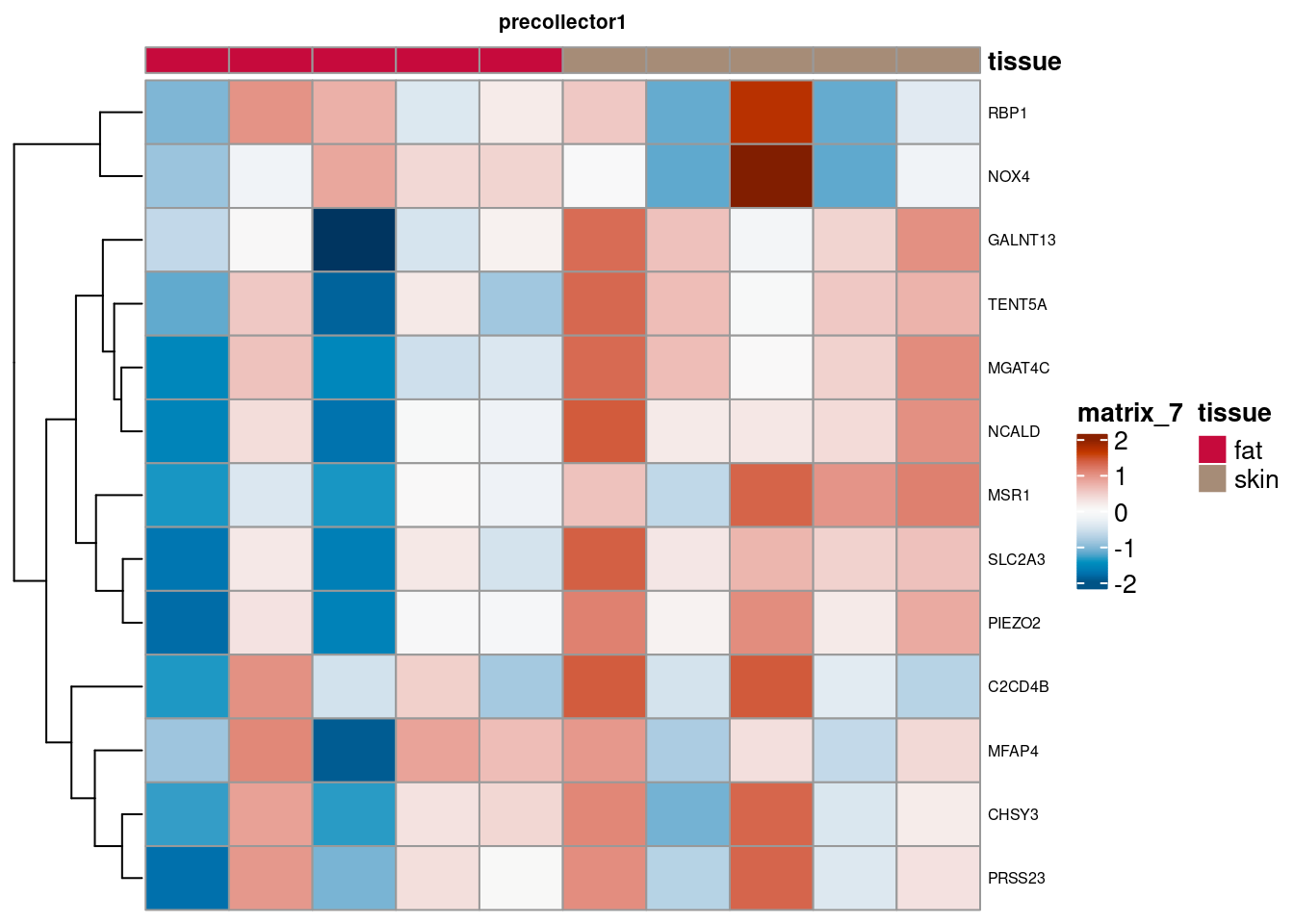
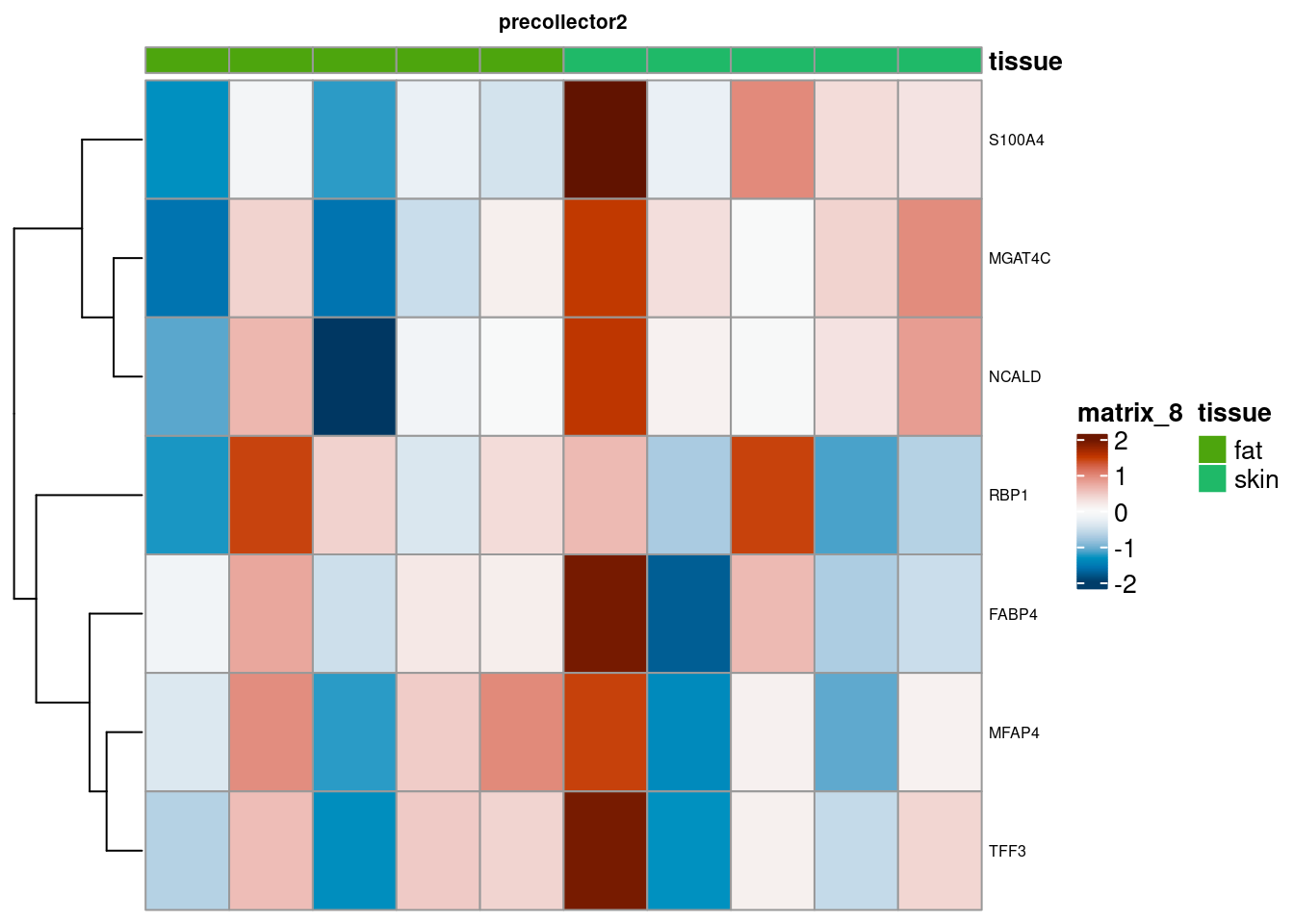
for (. in names(de_gen_list)) {
sub <- summed[,colData(summed)$ct_broad %in% ct_nam[[.]]]
sub <- sub[,order(sub$tissue)]
mtx_sub <- log1p(assays(sub)[["counts"]][rownames(de_gen_list[[.]]),])
colnames(mtx_sub) <- paste0("pseudobulk_", seq_along(1:ncol(sub)))
cd <- data.frame("tissue" = sub$tissue)
rownames(cd) <- colnames(mtx_sub)
hm <- pheatmap(mtx_sub,
main = ., fontsize = 6,
col = rev(hcl.colors(51, "RdBu")),
scale = "row",
show_colnames = FALSE,
cluster_cols = FALSE,
annotation_col = cd)
cat("####", ., "\n"); draw(hm); cat("\n\n")
}
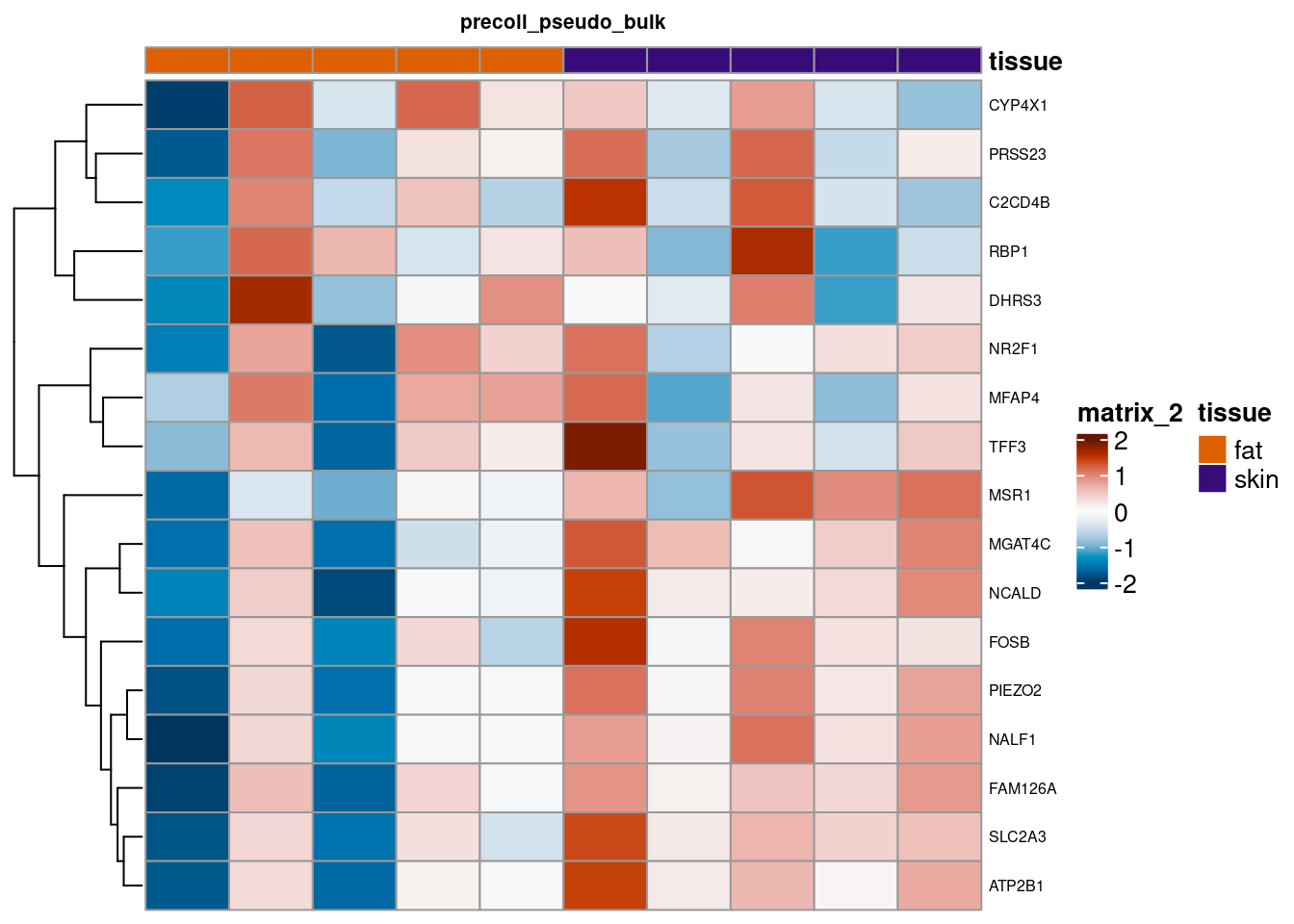
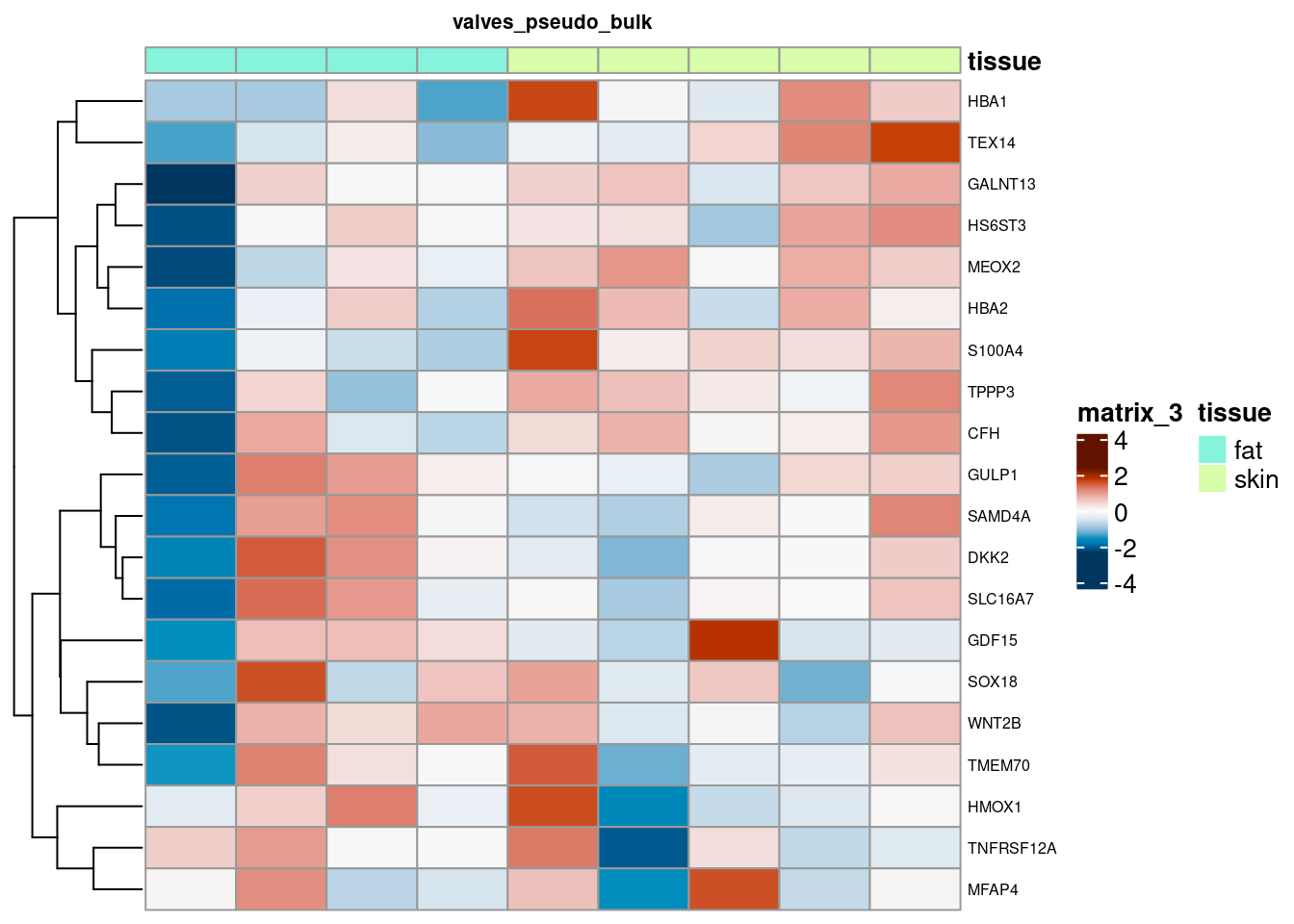
summed_all <- aggregateAcrossCells(sce,
id=colData(sce)[,c("tissue", "donor")],
use.assay.type = "counts")
summed_all <- summed_all[,summed_all$ncells >= 10]
rownames(summed_all) <- gsub("^.*\\.","",rownames(summed_all))
# subset res to de-genes
de_all <- as.data.frame(pb_all) |>
filter(FDR < 0.01) |>
slice_max(abs(logFC), n = 20)
summed_all <- summed_all[,order(summed_all$tissue)]
mtx_all <- log1p(counts(summed_all)[rownames(de_all),])
colnames(mtx_all) <- paste0("pseudobulk_", seq_along(1:ncol(summed_all)))
cd <- data.frame("tissue" = summed_all$tissue,
"donor" = summed_all$donor)
rownames(cd) <- colnames(mtx_all)
hm <- pheatmap(mtx_all,
main = "all", fontsize = 6,
col = rev(hcl.colors(51, "RdBu")),
scale = "row",
show_colnames = FALSE,
cluster_cols = FALSE,
annotation_col = cd)
draw(hm)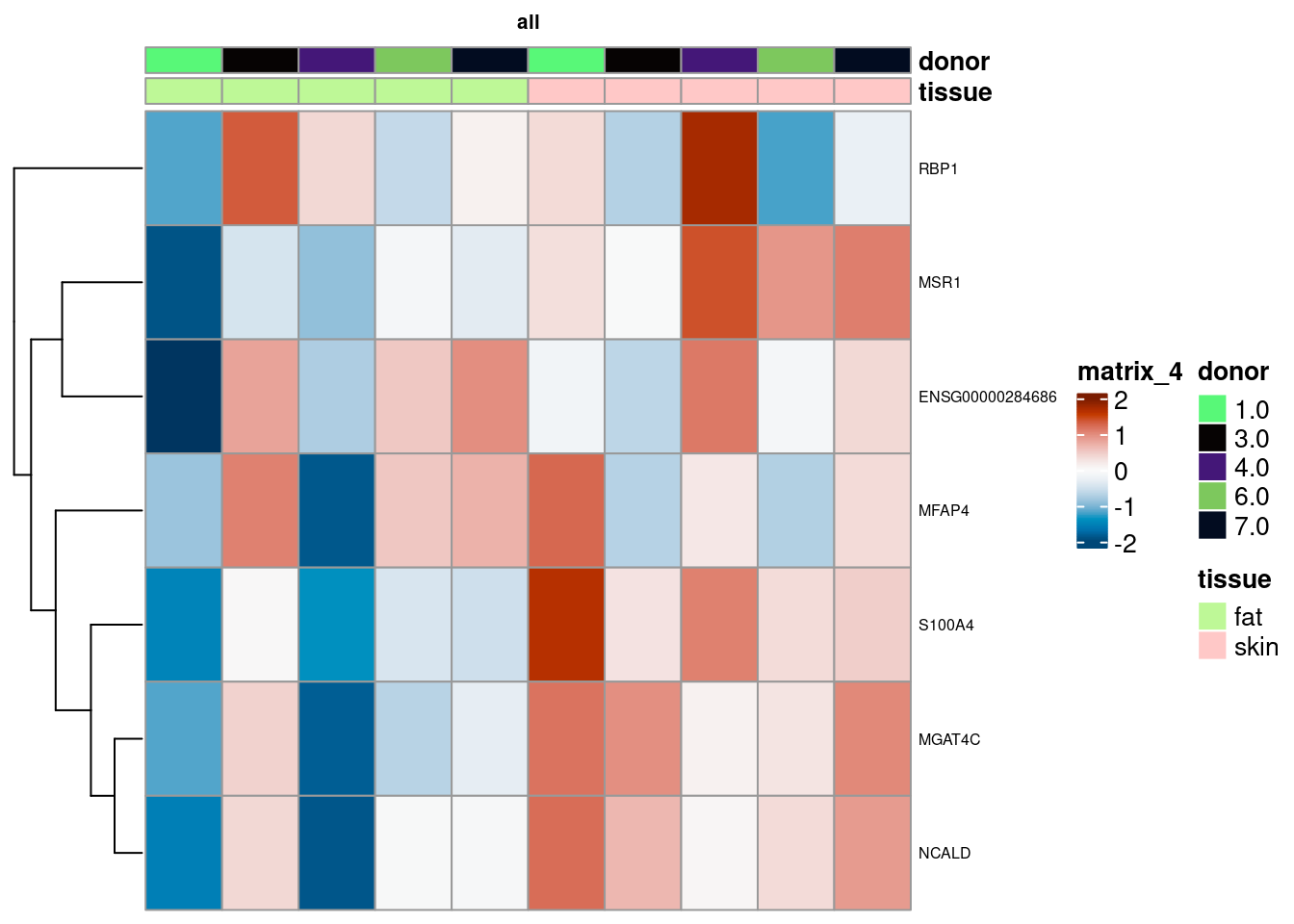
PB Heatmap subtypes
UpSet plot
upset_list <- lapply(pb_de, function(de_res){
de_res <- de_res |>
filter(FDR < 0.05 & abs(logFC) > 0.75)
rownames(de_res)
})
upset(fromList(upset_list), order.by = "freq")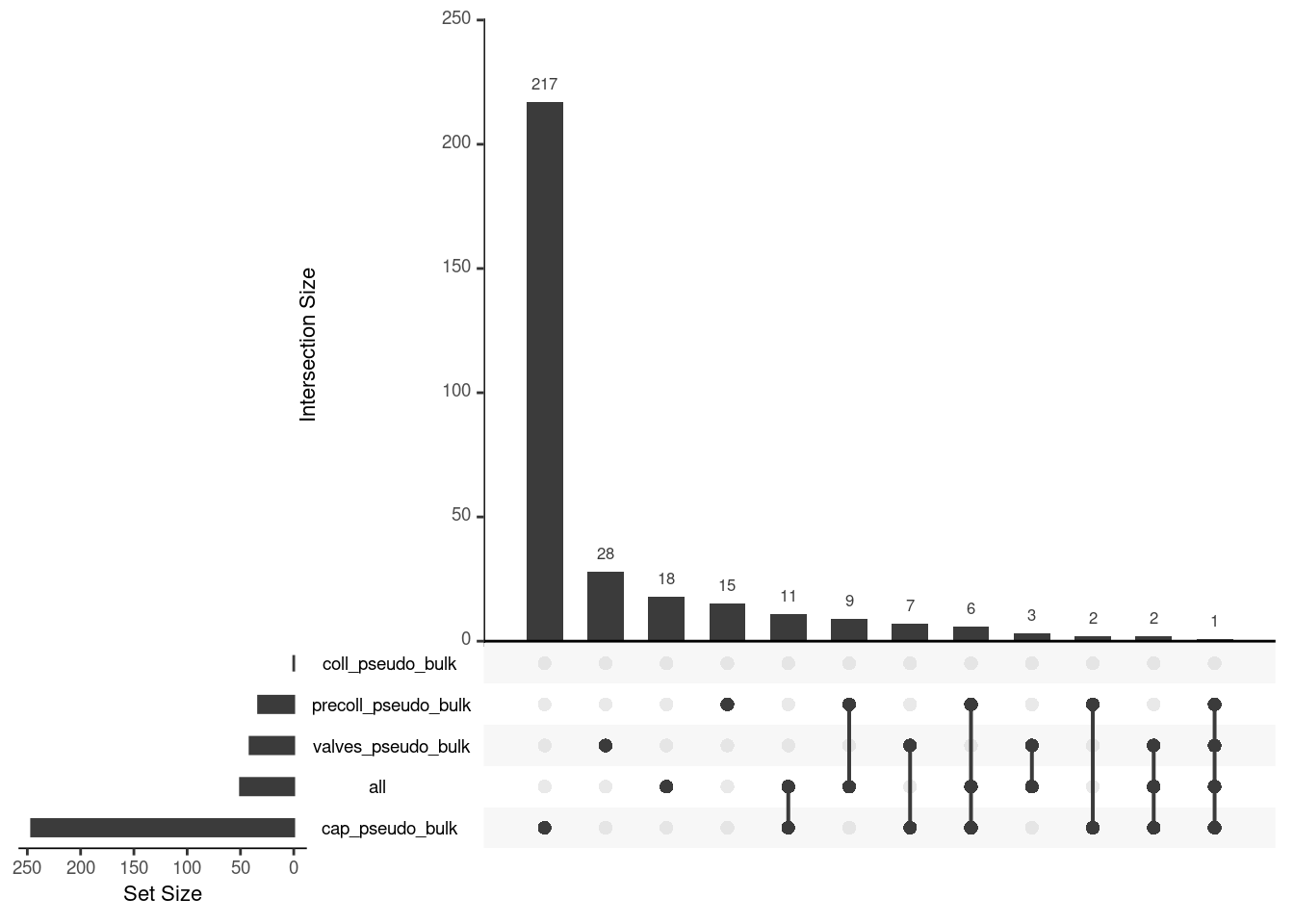
upset all de
upset_list <- lapply(pb_de, function(de_res){
de_res <- de_res |>
filter(FDR < 0.05)
rownames(de_res)
})
upset(fromList(upset_list), order.by = "freq")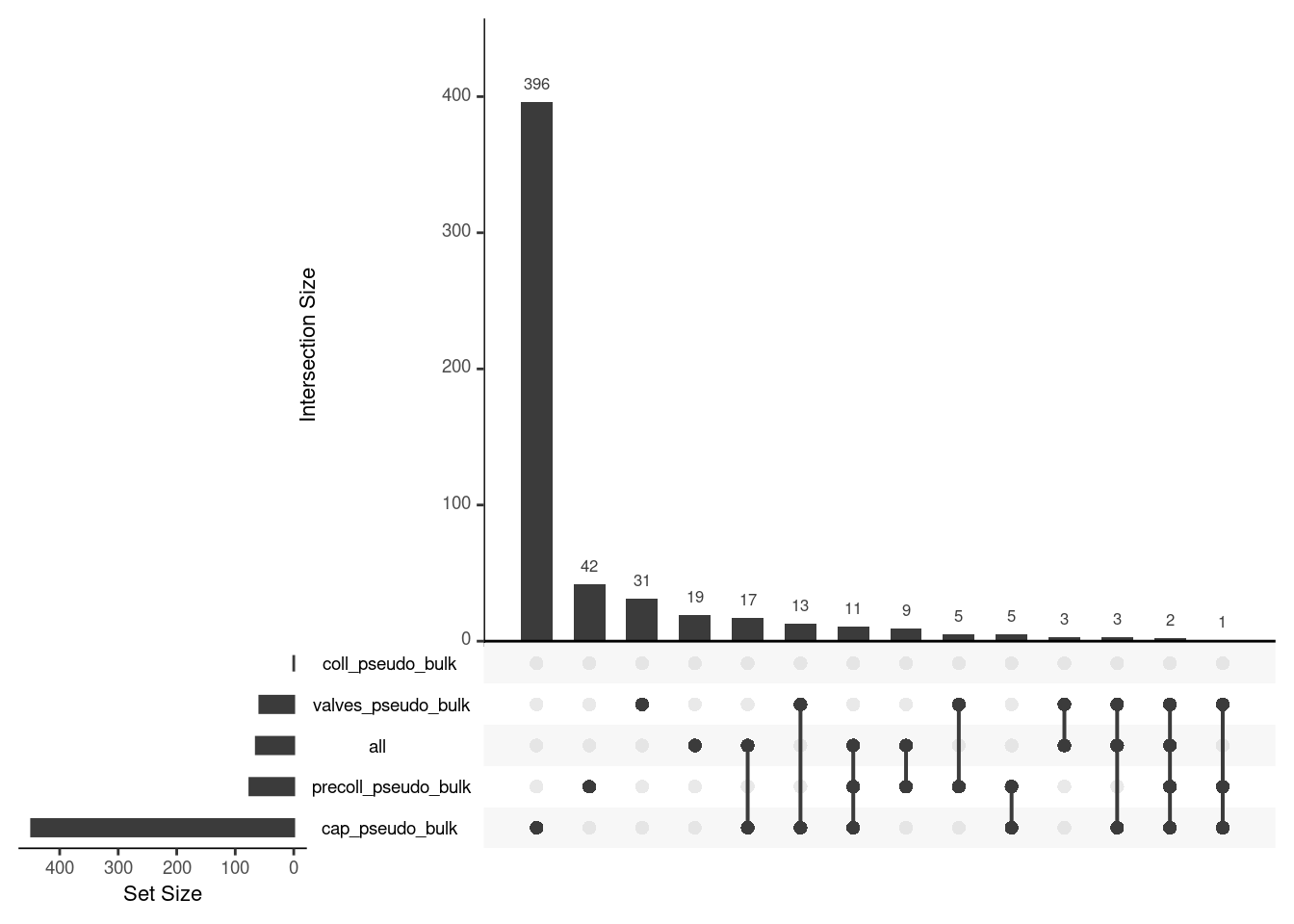
Boxplots DE genes
rownames(sce) <- gsub("^.*\\.","",rownames(sce))
sce_col <- sce[,sce$ct_broad %in% "collector"]
sce_col$ct_broad <- sce_col$ct_broad |> droplevels()
summed_col <- summed[,summed$ct_broad %in% "collector"]
summed_col$ct_broad <- summed_col$ct_broad |> droplevels()
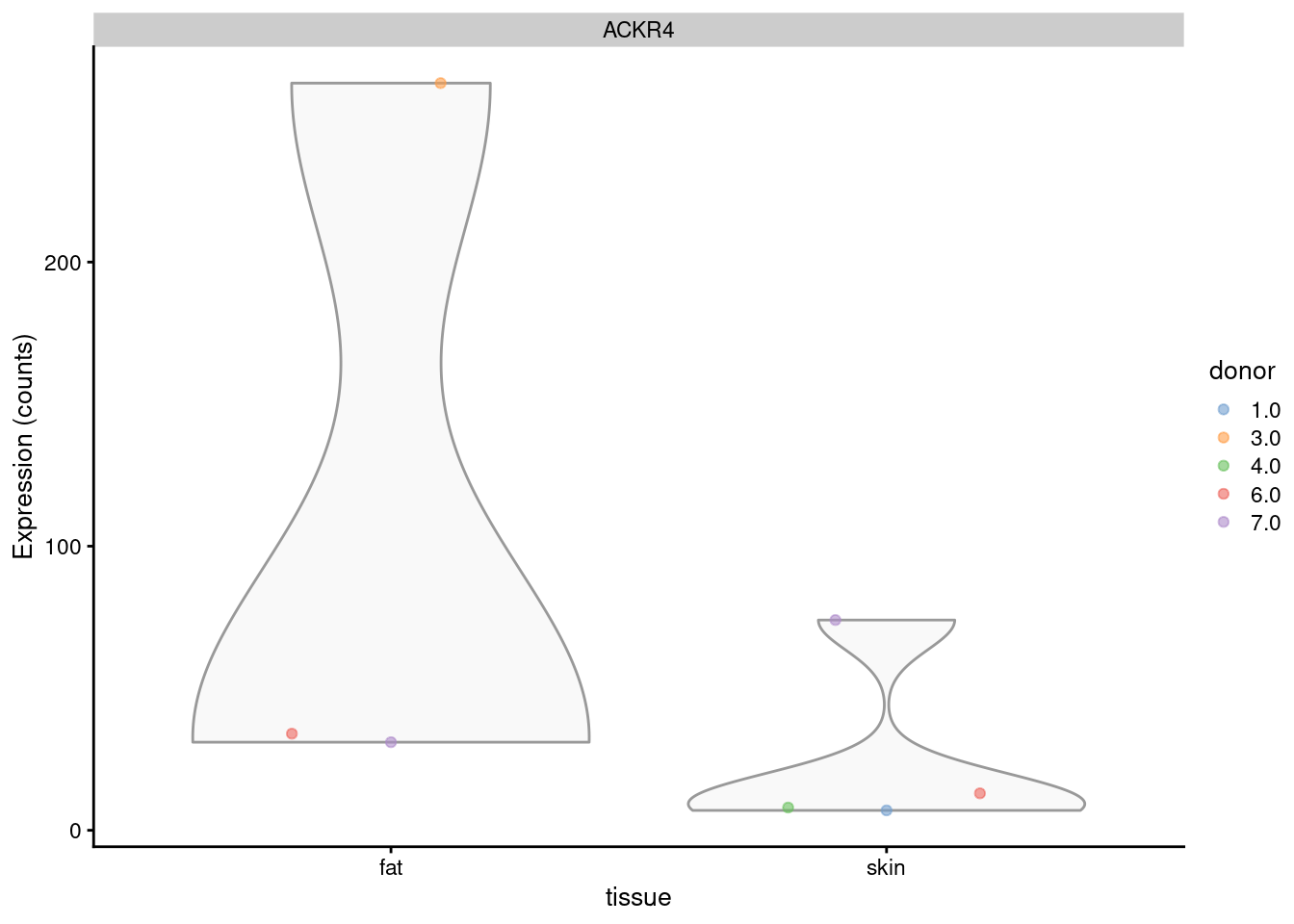
plotExpression(sce_col, features="ACKR4",
x="tissue", colour_by="donor")
plotExpression(summed_col, features="ACKR4",
x="tissue", colour_by="donor", exprs_values = "counts")